

Translate this page into:
A Case of Hearing Impairment with Renal Dysfunction
Address for correspondence: Dr. Sagar C. Kulkarni, Manipal Hospitals, Old Airpot Road, Bengaluru - 560 017, Karnataka, India. E-mail: sagarkulkarninephro@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Hearing impairment in a patient with renal failure is an important clue towards etiologic diagnosis of kidney disease. Variety of hereditary diseases, developmental defects, and toxins involve these two organs. However, additional retinopathy is seen in quite a few diseases which include Alport's syndrome and Muckle–Wells syndrome (MWS). We are reporting a case of middle-aged woman with childhood-onset of hearing impairment who presented with renal failure and was diagnosed to have renal amyloidosis on kidney biopsy but without any light chain restriction. During evaluation for live donor kidney transplant, her brother was also found to have hearing impairment and retinopathy however with normal renal function and urinalysis. Genetic testing of both of them was done for panel of mutations related to hereditary amyloidosis which revealed NLRP3 mutation in both. This mutation is characteristic of MWS which can lead to secondary amyloidosis and renal failure.
Keywords
Cryopyrin associated periodic syndrome
Muckle–Wells syndrome
Renal amyloidosis

Introduction
The association of hearing and renal impairment has been well recognized, but diagnosis of underlying problem is not always straight forward. Additional eye involvement points toward Alport's syndrome usually. Here we are reporting a case with hearing impairment, renal failure, and retinopathy due to amyloidosis secondary to Muckle–Wells syndrome (MWS). It is one of the entities under the category of cryopyrin-associated periodic syndrome (CAPS), which is a hereditary febrile syndrome with autosomal dominant inheritance. It is a rare disease mostly reported in Caucasian population. So far only 4 cases have been reported from Indian population.[1] Newer advances in molecular targeted therapies have suggested utility of IL-1 inhibitors to have therapeutic value. Hence, appropriate diagnosis of condition is important. In addition, screening of family members and consideration of therapeutic options might be useful.
Case Report
A 46-year-old woman presented to hospital with complaints of nausea, vomiting, and generalized weakness since 2–3 weeks associated with swelling over body. On evaluation, she was found to have severe renal dysfunction and initiated on hemodialysis. Her urine examination revealed 1+ protein and 3-4 RBCs with urine PCR of 2.48. She was continued on maintenance hemodialysis elsewhere after placement of left-arm AV fistula.
She came to our hospital for renal transplantation with younger brother being prospective donor. On evaluation, both of them had history of hearing impairment since childhood. Detailed family history revealed history of hearing impairment in her father and another younger brother Figure 1. Further investigations revealed sensorineural hearing loss and dot and fleck retinopathy in both of them. However, brother did not have proteinuria and/or renal dysfunction. Neither of them had any history of hematuria. In view of hereditary hearing impairment, retinopathy, and renal failure, possibility of Alport's syndrome was considered. Kidney biopsy was done which revealed nodular glomerulosclerosis with congophilic deposits in mesangium and arterioles which displayed apple-green birefringence on polarized light. IF was negative and kappa/Lambda did not show any restriction. Considering possibility of hereditary amyloidosis, genetic analysis was done which revealed mutation in NLRP3 gene in both patient and her brother. The variant identified was c.1049 C>T p.Thr350Met. This variant was found to be likely pathogenic for MWS, as per ACMG 2015 guidelines.[2] This variant has also been reported by Kuemmerle-Deschner et al.[3] in the cases of MWS. On retrospective enquiry, she also gave the history of episodes of cold-induced skin rash, arthralgia, and low-grade fevers. Hence, diagnosis of MWS was established. We offered genetic testing for other family members, but they have not consented for it.

- Pedigree chart for the presence of early-onset sensorineural hearing loss. Green: unaffected members; Red: affected members. Asterisk (*): individuals sequenced for genetic mutation
Discussion
MWS is a part of continuum of diseases with CAPS which include familial cold-induced autoinflammatory syndrome and neonatal-onset multisystem inflammatory disease [Figure 2]. CAPS is found predominantly in Caucasian population. Estimated prevalence in France was 1/360 000.[4] It has rarely been reported in non-Caucasian population.
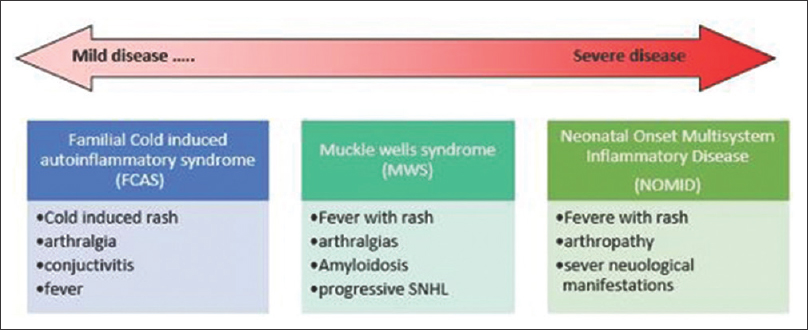
- Clinical spectrum of cryopyrin-associated periodic syndrome
The MWS is an intermediate phenotype characterized by recurrent episodes of fever, urticarial rash, arthralgia, myalgia, and conjunctivitis with progressive sensorineural hearing loss from childhood. Secondary amyloidosis type AA also develops later in life which can lead to renal failure. Up to 25% patients with MWS develop renal dysfunction. Mutation in NLRP 3 gene at chromosome 1q44 leads to excessive activation of caspases 1 and overproduction of Interleukin 1β.[5] This leads to formation of aberrant inflammasome. Interleukin 1β is responsible for chronic inflammation and consequent clinical manifestations. A recent European registry of 133 patients revealed 31 different NLRP3 mutations.[6] Of all mutations, T348M variant (which was found in our case), now known as T350M[3], was associated with early onset of disease, chronic course, and hearing loss.[6].
Clinical criteria have also been proposed for the diagnosis of CAPS by Kuemmerle-Deschner et al.[7] which are shown in Table 1.
| Mandatory criteria | Raised inflammatory markers (CRP*/SAA^) |
|---|---|
| Additional criteria - ≥2 of 6 CAPS typical signs/symptoms | 1.Urticaria-like rash |
| 2. Cold/stress-triggered episodes | |
| 3. Sensorineural hearing loss | |
| 4.Musculoskeletal symptoms (arthralgia/arthritis/myalgia) | |
| 5. Chronic aseptic meningitis | |
| 6. Skeletal abnormalities (epiphyseal overgrowth/frontal bossing) |
* -: C - reactive protein. ^ -: Serum Amyloid A.
Before introduction of IL-1 inhibitors, NSAIDS and antihistaminics were mainstay of treatment and steroids had only modest benefit. Three IL 1 inhibitors are currently available, viz., Anakinra, Rilonacept, and Canakimumab. Anakinra is an IL-1 receptor antagonist and Rilonacept is a fusion protein bearing IL-1 binding domains of IL-1 receptor, while Canakimumab is fully human IgG antibody against IL-1.[8] Biologicals have reported to improve quality of life significantly. However, they are expensive and have inherent side effect. It is reported that[8] biologicals should be used in patients presenting with severe phenotypes, those at risk of complications and those with persistent high CRP and/or SAA (Serum amyloid A), before development of ESRD. Scarpioni et al.[8] reported that 2 Caucasian patients with MWS treated with 60 doses of Canakimumab over 18 months resulted in significant improvement in glomerular filtration rate and proteinuria. SAA levels also normalized with treatment.
A thorough evaluation of cause of secondary amyloidosis is important in arriving at diagnosis. Considering recent advancements in treatment modalities and potential effect on altering natural course of disease, it is worthwhile to have high index of suspicion for CAPS in patients with hereditary fever, rash, and sensorineural hearing loss. Also, active screening of family members can identify at-risk persons and Potential treatment can be offered to prevent irreversible damage.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Muckle-Wells syndrome in an Indian family associated with NLRP3 mutation. J Postgrad Med. 2015;61:120-2.
- [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med. 2015;17:405-23.
- [Google Scholar]
- Early detection of sensorineural hearing loss in Muckle-Wells-syndrome. PediatrRheumatol Online J. 2015;13:43.
- [Google Scholar]
- Mutations in the autoinflammatorycryopyrin-associated periodic syndrome gene: Epidemiological study and lessons from eight years of genetic analysis in France. Ann Rheum Dis. 2011;70:495-9. doi: 10.1136/ard. 2010.138420 Epub 2010 Nov 24. Erratum in: Ann Rheum Dis 2012;71:1264
- [Google Scholar]
- Muckle-Wells syndrome: Clinical perspectives? Open Access Rheumatol. 2017;9:123-9. doi: 10.2147/OARRR.S114447 eCollection 2017. Review
- [Google Scholar]
- Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: A series of 136 patients from the Eurofever Registry. Ann Rheum Dis. 2015;74:2043-9. doi: 10.1136/annrheumdis-2013-204991
- [Google Scholar]
- Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS) Ann Rheum Dis. 2017;76:942-7.
- [Google Scholar]
- Renal involvement in secondary amyloidosis of Muckle-Wells syndrome: marked improvement of renal function and reduction of proteinuria after therapy with human anti-interleukin-1β monoclonal antibody canakinumab. Clin Rheumatol. 2015;34:1311-6.
- [Google Scholar]




