

Translate this page into:
A Rare Case of APRT Deficiency with End-stage Renal Failure and Successful Renal Transplant
Address for correspondence: Dr. Tarun K. Jeloka, Department of Nephrology, Aditya Birla Memorial Hospital, Pune - 411 033, Maharashtra, India. E-mail: tjeloka@yahoo.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Renal calculus disease is a common cause of renal injury. However, crystal nephropathy (uric acid, oxalate, and dihydroxyadenine) can present as chronic kidney disease without any evidence of renal stones. If left undiagnosed, there is a potential chance of recurrence in the allograft leading to graft failure after transplantation. Pretransplant identification and management can avoid such complications. Here, we describe a case of APRT deficiency leading to crystal nephropathy and end-stage renal failure in a patient who underwent a successful kidney transplant.
Keywords
APRT deficiency
crystal nephropathy
recurrence posttransplant

Introduction
Adenine phosphoribosyl transferase (APRT) enzyme deficiency is a rare inborn error of metabolism which results in excess of 2,8 dihydroxyadenine (DHA). DHA is excreted by the kidneys and precipitates in the urine leading to kidney stones and crystal nephropathy. If untreated, it leads to end-stage kidney disease (ESKD). There is a risk of recurrence of crystal nephropathy leading to graft loss after renal transplantation if the pretransplant diagnosis is unavailable and hence not treated. Therefore, diagnosis before transplantation and lifelong treatment after transplantation are important in preventing the risk of recurrence after renal transplant. Here, we present a rare case report of APRT enzyme deficiency leading to ESKD and successful renal transplantation.
Case Report
A 34-year-old male from a nonconsanguineous marriage and, without any medical history of morbidity, presented in August 2015 at an alternate center with progressive loss of appetite, nausea, and weakness for a few months. There were no complaints of swelling, oliguria, hematuria, breathlessness, or fatigue. He was evaluated and was found to have severe renal impairment. Kidney biopsy was done which showed features of chronic tubulointerstitial nephritis with pigmented crystals [Figure 1 and 2]. There were 12 glomeruli, which appeared normal with many tubules having pigmented yellow to brown, radially arranged crypts obliterating the tubular lumina. Pigments were birefringent under polarized light and black on silver stain. There was associated tubular injury with histiocytes lining the crystals in few areas. Tubular atrophy was seen in 20–25% of the cortex sampled. There was diffuse interstitial lymphocytic infiltrate. These features were supportive of 2,8 dihydroxy adeninuria with associated significant tubular injury, tubular atrophy, and diffuse interstitial inflammation. The crystals were pigmented and hence unlikely to be hyperoxaluria.

- Kidney biopsy showing pigmented dihydroxyadenine crystals
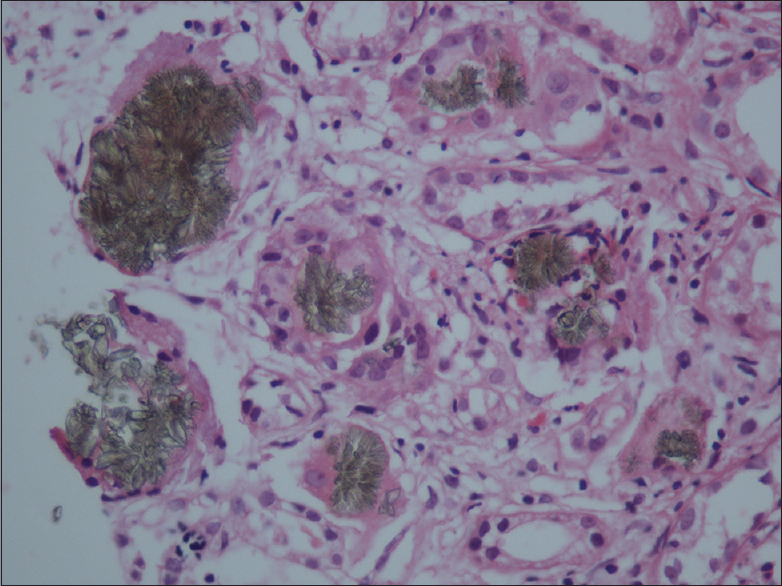
- Kidney biopsy showing pigmented dihydroxyadenine crystals (higher magnification)
There was no past or family history of renal calculus disease. APRT enzyme activity in RBCs was desired but was not available. Since he wanted a renal transplant, it was essential to rule out genetic causes of calculus disease such as oxalosis. Hence for confirmation, genetic testing was done for mutation analysis of genes (APRT and HOGA1) associated with renal stones (DHA and oxalate, respectively). A next-generation sequencing (NGS) panel (Illumina TruSight One), consisting of >4800 genes was run on DNA extracted from peripheral blood, using the Illumina MiSeq platform. Bioinformatic analysis was carried out on the coding regions of the APRT and HOGA1 genes. It revealed the presence of two clinically relevant heterozygous mutation variants c.3G>C (p.Met) and c.227C>T (p.Ala76Val) in the APRT gene. This is consistent with diagnosis of APRT deficiency leading to 2,8 DHA urolithiasis. The first variant c.3G>C is a single nucleotide change, present in the start codon of the APRT gene, and results in the loss of first amino acid methionine. A possible effect of this mutation is the creation of an alternate downstream translation initiation site with a new reading frame. This variant is annotated in the human gene mutation database (HGMD) as a known disease-causing mutation. The second variant is a novel single nucleotide variant, present in exon 3 of APRT gene and has been predicted to be pathogenic by various in silico analyses algorithms including Mutation Taster and PolyPhen. This variant has not been reported in the ExAC and 1000 genome databases, to date. But, based on available pieces of evidence in databases and prediction analysis, this variant was annotated as a “variant of unknown significance, likely pathogenic.” The pedigree was not investigated as family refused genetic testing.
He was on medical management with febuxostat till January 2016, when he was started and managed on maintenance hemodialysis. His renal transplant surgery was done on 29 July 2016 with his aunt as a donor. Posttransplant, he had delayed graft function and was discharged with 1.7 mg/dl creatinine. He was continued on febuxostat. Posttransplant, he was stable without any complaints with creatinine of 1.6 mg/dl on the last follow-up in March 2019.
Discussion and Review of Literature
The APRT enzyme is expressed in all tissues and is the only pathway for metabolic salvage of adenine resulting from polyamine biosynthesis and dietary sources.[1] In APRT deficiency, adenine is converted to 8-hydroxyadenine, which is further metabolized to DHA by xanthine dehydrogenase (XDH).[2] DHA has a high renal clearance, which may involve both filtration and tubular secretion. APRT deficiency, therefore, results in high urinary levels of DHA.[3] DHA is extremely insoluble in urine and forms crystals, which can gather, grow, and form stones or precipitate in the renal parenchyma, causing crystalline nephropathy.[45] APRT gene located on chromosome 16q24 encompasses 2.8 kb of DNA, containing five exons, and has a coding region of 540 bp.[6] The mode of inheritance of APRT deficiency is autosomal recessive.
There are 2 types of APRT deficiency. Manifestations of the disease are identical in both two types.[78910] APRT deficiency is often considered a very rare disease. Its worldwide prevalence remains largely unknown. Most cases and studies are published from Japan and are rarely reported from other populations except for Icelandic and French cohorts.[710].
Type I APRT deficiency: No enzyme activity. Mainly affects white individuals but has also been reported in people originating from countries worldwide.[7] Type II APRT deficiency: APRT activity is 25% of normal value[8] with reduced affinity for co-substrate 5-phosphoribosyl 1 pyrophosphate and most cases are reported from Japan[9].
Clinical presentation
APRT deficiency can present as urolithiasis and crystalline nephropathy. Although APRT deficiency occurs in all cells, no extra-renal symptom has been reported in affected individuals. APRT deficiency can present at any age from infancy to older than 70 years.[7910] In the setting of familial screening, it is not unusual to detect complete APRT deficiency in individuals who are totally asymptomatic even as adults.[710] The first urolithiasis episode may occur during the first few months after childbirth as well as later than the fifth decade.[7899] Reddish-brown diaper stains may be observed in infants.[10] Delayed diagnosis is less frequent when symptoms begin to show in children. In adults, confusion occurs between DHA and uric acid stones, which are radiolucent such as DHA stones. DHA stones may contain calcium salts, making them sometimes radiopaque.[11].
DHA crystals can precipitate in renal tubules and interstitium and severely impair renal function leading to crystalline nephropathy known as DHA nephropathy.[12] DHA nephropathy can present as an acute disease leading to renal failure (RF) in a few days or weeks or more frequently develop insidious and progressive decline of renal function in several years. Acute RF may be triggered by dehydration, which causes oliguria, urine supersaturation, and massive precipitation of DHA. Decreased renal function was observed in about one-third of patients at the time of diagnosis. Nearly 10% of patients had reached ESKD before diagnosis.[7910] DHA nephropathy can recur after renal transplantation if APRT deficiency remains unrecognized, which often leads to loss of allograft function within a few weeks.[51013] Factors such as fluids and purine intake are likely to account for variability in presentation.
Earlier reported cases of APRT deficiency from India include two cases in early childhood and one case in a young female.[141516] Both children (3 years and 2 years) presented with radiolucent stone with acute kidney injury in one and urinary tract infection in another. The only adult reported case was in a 24-year-old female, who presented with crystal nephropathy and rapidly progressive RF but had no stones. Our case also had crystal nephropathy without stones but was detected late in chronic kidney disease stage, who underwent transplant, which is reported for the first time.
Diagnosis
Stones should be analyzed by the combination of morphologic examination under a stereomicroscope and infrared spectroscopy which allows identification of DHA in all cases.[17] The biochemical stone analysis fails to differentiate DHA from uric acid and cannot be relied on for the diagnosis of APRT deficiency. Crystalluria examination by light and polarizing microscopy is a very useful, noninvasive, and inexpensive tool.[17] It is best performed in the first voided morning urine sample. Quantification can be done by counting the number of crystals per volume unit, which is high in untreated patients.[17] Infrared spectrophotometry should also be performed when DHA crystals are suspected. Crystalluria is very sensitive and specific, allowing the identification of homozygotes in 100% of cases. False-negative results have been rarely reported.[10].
As an invasive procedure, renal biopsy is theoretically not required. However, it often leads to the diagnosis in cases of DHA nephropathy as an unexpected finding in most cases.[13] Our case also was diagnosed following clues from the renal biopsy. The renal biopsy usually shows severe tubular injury and precipitation of crystals within tubular lumen and in renal interstitium. The best method is to characterize crystals in renal biopsy using polarizing microscopy and Fourier transform infrared microscopy.[18] If latter is not available, crystalluria and measurement of APRT activity assay may lead to diagnosis.
Measurement of APRT activity in erythrocyte lysates can be done by high-performance liquid chromatography (HPLC) and urinary adenine can be measured by UV detection.[19] APRT enzymatic activity is not mandatory but is useful when available. The mutations involved may be determined by the sequencing of PCR-amplified DNA which can be readily performed given the small size of APRT gene.[6] However, 10% of mutations remain unidentified.[7].
The possibility of APRT deficiency should be considered in
-
All cases of urolithiasis in children,
-
Recurrent urolithiasis (especially if stones are radiolucent),
-
Urolithiasis associated with renal dysfunction of uncertain cause.
In such cases, adequate stone analysis should be performed whenever possible. Crystalluria is very useful, especially if the stone is not available for analysis.[13] APRT activity assay is useful when no stone is available and crystalluria cannot be studied in a patient with crystalline nephropathy (e.g., anuria). APRT activity should be systematically measured in patients with ESRD awaiting renal transplantation who have a history of urolithiasis.[13] We could not do APRT enzyme activity, as this test is not available as of now in any part of the country. But, with the biopsy picture and pathogenic mutation identified on NGS, it is highly likely to be diagnostic of APRT deficiency.
Management
No treatment has been shown to increase APRT activity.
-
Allopurinol: It acts by blocking xanthine dehydrogenase. The possibility of renal function recovery depends on the degree of acute tubular necrosis and chronic tubulointerstitial changes when treatment is initiated. Complete APRT deficiency even asymptomatic should be treated by allopurinol, given the risk for acute or insidious DHA nephropathy. The usual dosage of allopurinol is 200–300 mg/day in adults and 5–10 mg/kg/day in children. The treatment is monitored by clinical evaluation and urine microscopy. The absence of urinary DHA crystals is indicative of adequate therapy
-
Febuxostat: Another xanthine dehydrogenase inhibitor represents a potential alternative in allopurinol intolerant patients.20 The usual dose is 80 mg/day
-
High fluid intake of at least 2.5 L of water per day in adults
-
Avoidance of purine-rich food should also be advised. Alkalization does not have to be recommended because DHA remains very insoluble at pH values below 8.5. Decrease of crystalluria usually should be considered as an adequate response to treatment.[7] Stones may recur in a minority of patients despite treatment.[7] Lack of adherence to treatment or insufficient dosing of allopurinol should be suspected in such situations or if crystalluria persists
-
Surgical management of DHA kidney stones is the same as for the management of other types of stones including ESWL
-
Treatment of ESKD: Treatment of end-stage kidney disease remains the same as dialysis or kidney transplantation.
All individuals with APRT deficiency require treatment with allopurinol or febuxostat for at least 6 weeks prior to transplant surgery, whenever possible. The unrecognized disease may lead to graft failure because of the recurrence of crystal nephropathy. Lifelong therapy with allopurinol or febuxostat after kidney transplantation is required to prevent DHA crystal nephropathy in transplant kidney.
Surveillance
All individuals with APRT deficiency should be followed up every 6–12 months to monitor:
-
Kidney function,
-
Assess urinary excretion of DHA crystals,
-
Medications compliance,
-
Periodic ultrasound of kidney for new kidney stones.
Conclusion
APRT deficiency represents an example of a disease that can potentially lead to ESKD but can easily be treated by medications. As many patients of ESKD remain undiagnosed and labeled as possible chronic tubulointerstitial disease, it is pertinent to rule out APRT deficiency before transplant to prevent the risk of recurrence in transplanted kidneys by continuing lifelong treatment. There is also need to make diagnostic tools especially APRT activity assay readily available.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 5'Methylthioadenosine is the major source of adenine in human cells. Adv Exp Biol. 1984;165:83-8.
- [Google Scholar]
- Metabolic and Molecular Bases of Inherited Disease, 8th ed. Mcgraw-Hill: New York; 2001. p. :2571-84.
- [Google Scholar]
- Complete deficiency of adenine phosphoribosyltransferase: Report of a family. N Engl J Med. 1977;297:127-32.
- [Google Scholar]
- 2,8 Dihydroxyadeninuria: Laboratory diagnosis and therapy control. Urol Int. 1988;43:174-8.
- [Google Scholar]
- Adenine phosphoribosyltransferase deficiency and renal allograft dysfunction. Am J Kidney Dis. 2001;37:E37.
- [Google Scholar]
- Identification of a single missense mutation in the adenine phosphoribosyltransferase (APRT) gene from five Icelandic patients and a British patient. Am J Hum Genet. 1991;49:1306-11.
- [Google Scholar]
- Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. J Am Soc Nephrol. 2010;21:679-88.
- [Google Scholar]
- Common characteristics of mutant adenine phosphoribosyltransferases from four separate Japanese families with 2,8-dihydroxyadenine urolithiasis associated with partial enzyme deficiencies. Hum Genet. 1985;71:171-6.
- [Google Scholar]
- Genetic and clinical studies on 19 families with adenine phosphoribosyltransferase deficiencies. Hum Genet. 1987;75:163-8.
- [Google Scholar]
- Clinical features and genotype of adenine phosphoribosyltransferase deficiency in Iceland. Am J Kidney Dis. 2001;38:473-80.
- [Google Scholar]
- Adenine phosphoribosyltransferase deficiency. Clin J Am Soc Nephrol. 2012;7:1521-7.
- [Google Scholar]
- Recurrence of crystalline nephropathy after kidney transplantation in APRT deficiency and primary hyperoxaluria. Can J Kidney Health Dis. 2015;2:31.
- [Google Scholar]
- Unusual cause of crystalline nephropathy. Saudi J Kidney Dis Transpl. 2018;29:462-5.
- [Google Scholar]
- 2, 8 Dihydroxyadenine urolithiasis: A case report and review of literature. Indian J Nephrol. 2009;19:34-6.
- [Google Scholar]
- Dihydroxyadenine stone with adenine phosphoribosyltransferase deficiency: A case report. Indian J Urol. 2017;33:246-8.
- [Google Scholar]
- Clinical value of crystalluria and quantitative morphoconstitutional analysis of urinary calculi. Nephron Physiol. 2004;98:31-6.
- [Google Scholar]
- Fourier transform infrared microscopy identification of crystal deposits in tissues: Clinical importance in various pathologies. Am J Clin Pathol. 1996;105:576-82.
- [Google Scholar]
- Adenine phosphoribosyltransferase (APRT) deficiency: Identification of a novel nonsense mutation. BMC Nephrol. 2014;15:102-8.
- [Google Scholar]
- Febuxostat compared with allopurinol in patients with hyperuricemia in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450-61.
- [Google Scholar]




