

Translate this page into:
A Rare Genetic Mutation in a Stone Former
Address for correspondence: Dr. Periandavan Kalaiselvi, Department of Medical Biochemistry, Taramani Campus, University of Madras, Chennai - 600 113, Tamil Nadu, India. E-mail: pkalaiselvi2011@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
A 30-year-old woman with history of passage of stones since childhood presented with oliguria and pedal edema for 10 days. She had hypertension with a creatinine of 4.1 mg/dL. Evaluation showed presence of bilateral multiple renal calculi with features of chronicity of kidney disease. Metabolic work-up for nephrolithiasis turned out to be negative and eventually renal biopsy revealed features of chronic interstitial nephritis with greenish brown refractile crystals in the tubular lumen and interstitium. The possibility of dihydroxy adenine crystalline nephropathy was considered. Spectrophotometry of RBC lysates revealed decreased activity of Adenine phosphoribosyl-transferase enzyme. Gene amplification by PCR and sequential analysis identified a missense mutation in exon 3 region of APRT gene in the patient and her family members. This case report highlights the need to contemplate the diagnosis of DHA crystalline nephropathy in young patients with nephrolithiasis and the identification of a rare genetic mutation, which is being reported for the first time in India.
Keywords
Adenosine phosphoribosyl transferase
chronic kidney disease
2,8-Dihydroxyadenine nephropathy
genetic mutation

Introduction
Adenine phosphoribosyl-transferase enzyme (APRT) deficiency is a rare autosomal recessive inherited disorder of purine metabolism. In normal individuals, APRT enzyme metabolizes adenine to adenosine monophosphate. In its absence, adenine is metabolized by xanthine dehydrogenase (XDH) to 2,8-dihydroxyadenine (DHA), which is excreted in the urine. This DHA is insoluble in urine at the physiological range of pH resulting in crystalluria. Individuals differ in their ability to supersaturate the urine with DHA, thereby resulting in varied presentations even among homozygotes. The spectrum ranges from an asymptomatic state to recurrent nephrolithiasis leading onto chronic kidney disease (CKD). We describe a female patient with childhood onset stone disease, who developed chronic interstitial nephritis secondary to 2,8-DHA crystal deposition.
Case Report
A 30-year-old female, born of 2nd degree consanguineous marriage, was referred to our department with the complaints of pedal edema and oliguria associated with right loin pain for the past 10 days. She also had history of recurrent episodes of colicky abdominal pain with passage of stones since 7 years of age, for which she had not undergone any evaluation.
On examination, her blood pressure was 160/94 mm Hg. Dipstick urinalysis revealed hematuria of 3 (+) with no albuminuria and a urine pH of 6. Urine microscopy revealed 5–6 RBCs/hpf and was negative for crystals. Her hemoglobin was 9.2 g/dl, and white blood cell count was 10,300 cells/mm3. She had a creatinine of 4.1 mg/dl and potassium of 6.0 meq/L. Serum calcium was 7.9 mg/dl, albumin-3.7 g/dl, phosphorous –5.9 mg/dl, and uric acid- 9.7 mg/dl. Serum alkaline phosphatase was 236 IU/L, parathormone level - 167 pg/ml, and arterial blood gas analysis revealed high anion gap metabolic acidosis. On further evaluation, X-ray KUB was normal but ultrasonogram revealed right kidney of 8.6 × 3.9 cm size, with dilated pelvicalyceal system and a left kidney of 8.0 × 3.7 cm size, with multiple renal calculi.
A computed tomography (CT) scan showed left kidney with multiple renal calculi and small calculi in the pelvicalyceal system. A calculus of 8 × 5 mm was present at the interpolar region of right kidney along with an upper ureteric calculus. The right pelvicalyceal system was dilated with a thinned-out cortex. Also, a microlith was present near the right vesico-ureteric junction. She underwent bilateral ureteric stenting. Post-procedure, her urine output improved to about 2 litres per day with resolution of pedal edema but her renal failure persisted with a best creatinine of 3.3 mg/dl. The 24-h urine biochemical analysis could not be done due to presence of significant renal failure.
The possible differentials of childhood onset kidney stone disease included cystinuria, dent disease, familial hypomagnesemia with hypercalciuria and nephrocalcinosis, primary hyperoxaluria, and APRT deficiency. Our patient was a female; urinalysis did not reveal any crystals; metabolic work-up was not contributory and features of nephrocalcinosis were absent in CT.
Hence, in-view of persisting unexplained renal failure, she underwent a renal biopsy from the left kidney, which revealed greenish brown refractile crystals with central spicules, seen within tubules and interstitium, surrounded by multinucleated foreign body type of giant cells. These crystals were birefringent under polarized light suggesting the possibility of 2,8 DHA crystals [Figure 1]. Interstitial fibrosis with tubular atrophy involved one-third of the core.

- ×400, Haematoxylin and eosin staining showing interstitial inflammation around intra-tubular, greenish brown DHA crystals that are birefringent under polarized light
Diagnosis of APRT enzyme deficiency was entertained since 2,8-DHA crystals are pathognomonic of this condition. APRT enzyme activity measured in red blood cell lysates of the patient using spectrophotometry revealed markedly decreased APRT activity [Table 1].
| Age | Sex | APRT activity (nmoles/mg Hb/h) | Genotypes | Mutation (Nucleotide changes) | |
|---|---|---|---|---|---|
| Patient | 30 | F | 5.93 | Homozygous | GG⟶AA |
| Patient’s Sister | 24 | F | 18.2 | Heterozygous | GG⟶GA |
| Patient’s Brother | 36 | M | 9.2 | Homozygous | GG⟶AA |
| Patient’s maternal uncle | 56 | M | 15.6 | Heterozygous | GG⟶GA |
| Patient’s Mother | 54 | F | 17.42 | Heterozygous | GG⟶GA |
Normal range for APRT activity is 15-35 nmoles/mg Hb/h
The patient and her family members were screened for APRT gene mutation. The human APRT gene was amplified by PCR and sequential analysis demonstrated a point mutation with conversion of G to A at nucleotide 200 in exon 3 region. This mutation caused a change from CGA to CAA leading to change from arginine to glutamine [Figure 2]. The patient was homozygous containing only the mutant alleles. Her mother, sister, and maternal uncle were heterozygous and acting as carriers, while her elder brother was also homozygous for the same mutation [Figure 3].
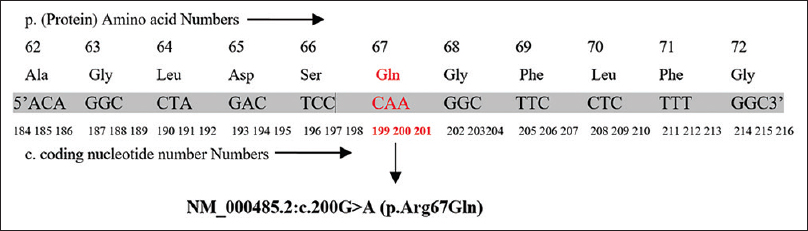
- The nucleotide sequence of the Exon 3 in the APRT allele showing a homozygous point mutation at codon 200 CGA to CAA (NM_000485.2:c. 200G>A)

- (a) Pedigree and mutational analysis of the proband (arrow) in the family. (b) Chromosome 16q24 Exon pattern of adenine phospho-ribosyl transferase (APRT) and the exon 3 amplified
She was advised low purine and low salt diet with liberal fluid intake. She was started on tablet Febuxostat 40 mg per day and was advised to continue the same lifelong. Currently, her serum creatinine is stable at 3.4 mg/dL and she is on follow-up on outpatient basis.
Discussion
APRT deficiency is a rare disorder of purine metabolism with autosomal recessive inheritance. In the absence of APRT enzyme, adenine is oxidized by XDH to 2,8-DHA, which is excreted in the urine. 2,8-DHA being poorly soluble at physiological pH results in crystalluria, recurrent nephrolithiasis, and less commonly, crystalline nephropathy.[1] Two types of APRT deficiency are described. Type I (complete deficiency in vivo and in vitro) is the most common type, predominantly affecting Caucasians. Type II (complete in vivo but 10-25% activity in vitro) has been described exclusively in the Japanese[123]
The APRT gene located on 16q24 is 2.6 kb long and contains 5 exons encoding 180 amino acids. Around 40 different mutations including missense, nonsense, insertion, deletion, and splicing have been reported. Despite sequencing the entire coding region and intron-exon junctions of the APRT gene, around 10% of mutations still remain undetermined.[3]
The commonly occurring mutations reported in literature are:
-
IVS4 + 2insT mutation, resulting in deletion of exon 4 during mRNA splicing, observed in European countries.
-
Met136Thr and g.442 T>C missense mutations in exon 5, G329A nonsense mutation in exon 3 and four-base pair (CCGA) duplication in exon 3, seen in Japanese patients.
-
A-to-T transversion in exon 3 (g.194A>T, p.Asp65Val) missense mutations, observed in British, Iceland, and Spanish populations.
-
p.Gln147X nonsense mutation in exon 5, identified in Italian patients.[34]
However, to the best of our knowledge, this is the first report from India to describe the presence of missense mutation 000485.2:c. 200 G > A (p.Arg67Gln), confirmed by sequential analysis of the entire family. This mutation led to conversion of G to A at nucleotide 200 in exon 3 region, causing change from CGA to CAA, resulting in an amino acid substitution from arginine to glutamine. The end result being, production of an abnormal non-functional APRT enzyme.
APRT deficiency eludes diagnosis due to the absence of disease-specific manifestations and lack of awareness of the disease. The age at diagnosis varies greatly, usually being unrecognized for years. Almost one-third slowly progress to CKD, with 10% of cases progressing to end-stage renal disease even before the diagnosis is made. This causes the disease to recur after renal transplantation, which can lead to allograft dysfunction within a few weeks.[5]
Kidney biopsy of our patient revealed chronic interstitial nephritis along with crystal deposition. The mechanism behind development of irreversible tubular atrophy and interstitial fibrosis due to DHA crystal deposition in the kidney is unclear. Studies showed APRT-deficient mice to have increased mRNA and gene expressions of MCP-1, IL-1β, CCR2, and TGF-β which was associated with interstitial macrophage infiltration and increased fibroblast activation. DHA crystals may possibly evoke this inflammatory response by stimulating tubular epithelial cells which leads onto fibrosis. Tubular obstruction by the intraluminal crystals may also be a factor in the development of renal damage.[67]
An intriguing finding in our report was that the patient's elder brother (36 years of age) was also homozygous for the same mutation, but did not manifest the disease. The reasons behind the variability in APRT deficiency phenotype are unclear and no correlation between phenotype and genotype has been reported. Factors like fluid intake, purine intake, and individual differences in the effectiveness of crystallization inhibitors like osteopontin may account for this variability.[8]
Crystalluria examination by light and polarizing microscopy is a sensitive and inexpensive tool for the identification of DHA crystals, when performed in first-voided morning urine samples. However, crystalluria may not be detected despite multiple urinalyses in patients with significant renal failure as in our patient, due to decreased 2,8-DHA clearance in these patients.[2] Occasionally, DHA crystals may be mistaken for uric acid stones as both of them are radiolucent and this misdiagnosis leads to treatment with allopurinol and low-purine diet, which are also effective in preventing DHA stone recurrence.[9] Differentiation between the two can be made only by ultraviolet or infrared spectrophotometry.[5]
DHA nephropathy needs to be distinguished from primary hyperoxaluria which also has a similar pattern of presentation. However, in contrast to primary hyperoxaluria, which is a systemic disease, DHA nephropathy does not appear to be associated with any extrarenal manifestations.[9] The gold standard for diagnosis is by measurement of APRT activity in red blood cells and genetic testing is confirmatory. Screening of siblings using urine microscopy for crystalluria and APRT enzyme assay is advocated in confirmed cases.[2]
The mainstay of treatment of DHA disease due to APRT deficiency includes a low-purine diet, high fluid intake and treatment with xanthine dehydrogenase inhibitors, either allopurinol or febuxostat, in order to effectively reduce the generation of 2,8-DHA. This prevents recurrent nephrolithiasis and can preserve renal function or even improve it, if initiated early enough.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
Dr. Anila Abraham Kurien – Director – Centre for Renal and Urological Pathology Private Limited reported the renal biopsy.
References
- Genetic and clinical studies on 19 families with adenine phosphoribosyltransferase deficiencies. Hum Genet. 1987;75:163-8.
- [Google Scholar]
- Clinical features and genotype of adenine phosphoribosyltransferase deficiency in Iceland. Am J Kidney Dis. 2001;38:473-80.
- [Google Scholar]
- Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. J Am Soc Nephrol. 2010;21:679-8.
- [Google Scholar]
- The metabolic and molecular bases of inherited disease. In: Scriver CR, Baudet AL, Sly WS, Valle D, eds. Adenine Phosphoribosyltransferase Deficiency and 2,8-Dihydroxyadenine Lithiasis. NY, USA: McGraw-Hill; 2001.
- [Google Scholar]
- 2, 8-dihydroxyadenine stone formation in a renal transplant due to adenine phosphoribosyltransferase deficiency. J Urol. 1996;156:1754-5.
- [Google Scholar]
- Adenine phosphoribosyltransferase-deficient mice develop 2, 8-dihydroxyadenine nephrolithiasis. Proc Natl Acad Sci. 1996;93:5307-12.
- [Google Scholar]
- Progressive renal dysfunction and macrophage infiltration in interstitial fibrosis in an adenine-induced tubulointerstitial nephritis mouse model. Histochem Cell Biol. 2009;131:483-90.
- [Google Scholar]
- Aprt/Opn double knockout mice: Osteopontin is a modifier of kidney stone disease severity. Kidney Int. 2005;68:938-47.
- [Google Scholar]
- Crystalline nephropathy due to 2, 8-dihydroxyadeninuria: An under-recognized cause of irreversible renal failure. Nephrol Dial Transplant. 2010;25:1909-15.
- [Google Scholar]




