

Translate this page into:
A Unique Case of Joubert Syndrome with Concurrent IgA Nephropathy and Nephronophthisis in an Adult Patient
Corresponding author: Mythri Shankar, Department of Nephrology, Institute of Nephro-Urology, Bangalore, Karnataka, India. E-mail: mythri.nish@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Shankar M, Anusha V, Shetty A, Sreedhara CG, Vinay KS, Aralapuram K, Reddy GG. A Unique Case of Joubert Syndrome with Concurrent IgA Nephropathy and Nephronophthisis in an Adult Patient. Indian J Nephrol. doi: 10.25259/IJN_86_2024
Abstract
A 30-year-old male born from a consanguineous marriage, with intellectual disability, developmental delay and Type 1 diabetes presented with polyuria and uremic symptoms. Physical examination revealed hypertension, retinitis pigmentosa, bilateral rotatory grade 3 nystagmus, eyelid droop, truncal obesity, acanthosis nigricans, and muscle hypotonia. Laboratory tests indicated kidney dysfunction. Magnetic resonance imaging of the brain showed the “molar tooth sign,” a hallmark of Joubert syndrome. The kidney biopsy highlighted features of IgA nephropathy, diabetic nephropathy, and nephronophthisis. Whole exome sequencing identified a homozygous nonsense variant in the AHI1 gene, known to cause Joubert syndrome 3. This case is unique due to its genetic proof of an AHI1 mutation causing Joubert syndrome in an Indian patient and the co-occurrence of IgA nephropathy with nephronophthisis.
Keywords
Jouberts syndrome
IgA nephropathy
Retinitis pigmentosa
Nystagmus
Genetic study

Introduction
Joubert syndrome is categorized within the ‘cerebello-oculo-renal syndromes’ and is an inherited condition. It is defined by the underdevelopment of the cerebellar vermis in the brain’s midline, a deeper interpeduncular fossa, and longer, thicker superior cerebellar peduncles, which create molar tooth sign (MTS) in imaging studies. Symptoms include muscle weakness, delays in development, and cognitive impairment. Marie Joubert and her team were the first to identify this syndrome in 1969.1
Case Report
A 30-year-old male with intellectual disability and developmental delay, type 1 diabetes since 12 years presented with nausea, vomiting, nocturia and polyuria. He was the eldest offspring from a third-degree consanguineous marriage. The diagnosis given was “cerebral palsy”. Upon physical assessment, he was found to be hypertensive, had retinitis pigmentosa, bilateral rotatory grade 3 nystagmus, eyelid drooping, truncal obesity, acanthosis nigricans and muscle hypotonia.
Investigations revealed anemia, kidney dysfunction and proteinuria (4.5g/day) [Table 1]. Magnetic resonance imaging of the brain displayed the distinct “molar tooth sign” [Figure 1]. Ultrasound revealed normal-sized kidneys with partially maintained cortico-medullary differentiation.
| Laboratory investigation | Results |
|---|---|
| Urine routine and microscopy | Protein 3+, RBC - 2-4/HPF, WBCs 4-6/HPF |
| 24-hour urine protein | 4.5g/5000ml |
| Hemoglobin (g/dl) | 9.5 |
| Total count (cells/mm3) | 8740 |
| Platelet count (cells/mm3) | 2.57 lakh |
| Blood urea (mg/dl) | 128 |
| Serum creatinine (mg/dl) | 5.18 |
| Serum sodium (mEq/L) | 134 |
| Serum potassium (mEq/L) | 4.3 |
| Serum chloride (mEq/L) | 110 |
| Serum bicarbonate (mEq/L) | 13 |
| Serum calcium (mg/dl) | 7.4 |
| Serum phosphorous (mg/dl) | 6.1 |
| Alkaline phosphatase (IU/L) | 150 |
| Serum uric acid (mg/dl) | 6.1 |
| Indirect bilirubin (mg/dl) | 0.3 |
| Direct bilirubin (mg/dl) | 0.6 |
| Serum protein (g/l) | 8.6 |
| Serum albumin (g/l) | 4 |
| Total cholesterol (mg/dl) | 204 |
| Low-density lipoprotein (mg/dl) | 125 |
| 2D ECHO | Hypertensive heart disease, mild left ventricular hypertrophy, left ventricular ejection fraction - 60% |
RBC - Red blood cell; WBC - White blood cell; HPF - High power field; ECHO – Echocardiogram
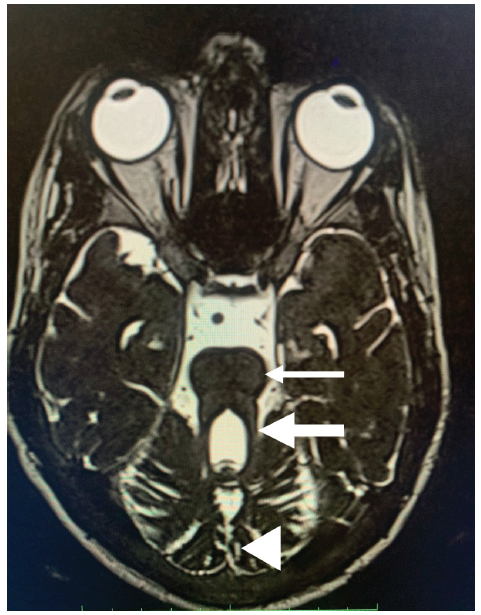
- Magnetic resonance imaging (MRI) brain T2 weighted image, axial section. The thin white arrow shows the “Molar Tooth Sign.” The thick white arrow shows an elongated cerebellar peduncle. The white arrowhead shows cerebellar vermis hypoplasia.
Kidney biopsy showed ten glomeruli of which six were globally sclerosed. Viable glomeruli showed mesangial expansion with increased matrix deposition. The capillary lumen was showed segmental thickening. Bowman’s capsule was thickened with periglomerular fibrosis. Tubules showed thickened basement membranes and tubular atrophy (about >50% of the cortex studied) [Figure 2]. Immunofluorescence showed granular mesangial positivity of IgA (+2/+3), and C3 (+2). Features were consistent with IgA nephropathy (Oxford scoring: M1 E0 SO T2 CO). Tubular basement membrane thickening and interstitial fibrosis were suggestive of background nephronophthisis. Glomerular basement membrane thickening and mesangial expansion were suggestive of diabetic nephropathy.
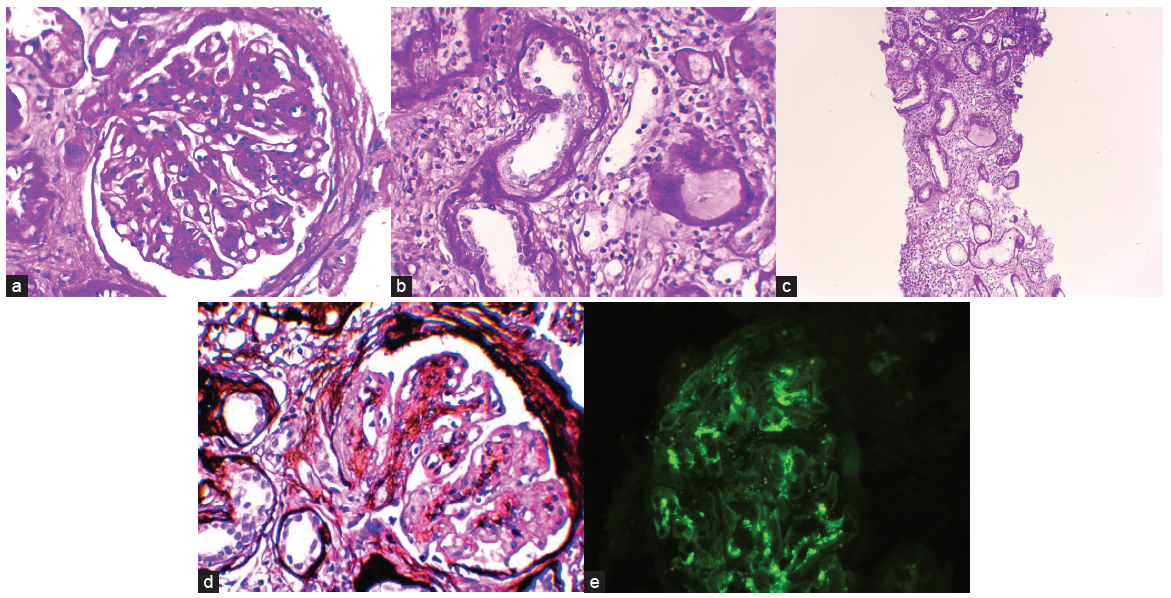
- (a) Light microscopy (100X): Mesangial expansion and hypercellularity. (b) Light microscopy (100X): Tubular basement membrane thickening. (c) Light microscopy (40X): Tubular basement membrane thickening and interstitial inflammation. (d) Light microscopy (Methenamine silver stain, 100X): Mesangial expansion and hypercellularity. (e) Immunofluorescence: Mesangial IgA deposits.
Whole exome sequencing revealed homozygous nonsense variant in Exon 9 of the AHI1 gene (chr6:g.135457660G>A; Depth: 41x) that results in a stop codon and premature truncation of the protein at codon 329 (p.Arg329Ter; ENST00000265602.11). The variant has been reported in patients with Joubert syndrome 32 and classified as pathogenic in the ClinVar database. He was started on conservative management for kidney disease.
Discussion
This case report is unique as it is the first genetically proven AHI1 mutation causing Joubert’s syndrome reported from India. The patient also has features of IgA nephropathy in kidney biopsy along with nephronophthisis, which is also the first case to be reported worldwide. It is unknown if the association was incidental or due to Joubert’s syndrome.
The incidence of Joubert Syndrome Related Disorders (JSRD) is estimated to range from 1 in 80,000 and 1 in 100,000 live births.3 These conditions are categorized as ciliopathies4 and are linked to mutations in more than 30 genes, with the AHI1, TMEM67, and CEP290 genes being among the most frequently affected.5 JSRD is divided into six phenotypic categories6 and display a wide range of clinical manifestations, merging neurological symptoms with the involvement of multiple organs such as the retina, kidneys, liver, and bones.7,8 In patients with Joubert syndrome linked to AHI1 mutations, approximately 75% experience retinal disorders.
Around 25% of individuals with JSRD experience kidney disease, often manifesting as nephronophthisis (NPH). NPH is identified by its distinctive changes in kidney structure, including an irregular and thickened basement membrane of the tubular epithelium and progressive scarring of kidney tissue. Juvenile NPH might not show symptoms for years, potentially presenting only subtle signs like increased urination and thirst until significant kidney dysfunction appears in late childhood or early adolescence that may progress to end-stage kidney disease. The identification of MTS with cystic dysplastic kidneys (CDK) led some researchers to propose a new condition named Dekaban-Arima syndrome. Recent reviews of cases initially classified under this syndrome have found histological signs more consistent with NPH than CDK, suggesting a uniform kidney disease phenotype across all JSRD.9 CDK also appears in other ciliopathies, like in the fatal Meckel syndrome.10
Identifying specific genetic mutations facilitates early prenatal genetic testing, even though fetal brain imaging may not provide clear results until late in the second trimester of pregnancy.6
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
References
- Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813-25.
- [CrossRef] [PubMed] [Google Scholar]
- DNA analysis of AHI1, NPHP1 and CYCLIN D1 in joubert syndrome patients from the netherlands. Eur J Med Genet. 2008;51:24-34.
- [CrossRef] [PubMed] [Google Scholar]
- Joubert Syndrome Foundation. 2003. Available from: http://www.joubertsyndrome.org] [Last accessed 2024 Feb 19].
- Joubert syndrome: Report of 11 cases. Turk J Pediatr. 2012;54:605-11.
- [PubMed] [PubMed Central] [Google Scholar]
- Mutation spectrum of joubert syndrome and related disorders among arabs. Hum Genome Var. 2014;1:14020. Erratum in: Hum Genome Var 2015;2:15001
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Joubert syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The primary cilium as a cellular signaling center: Lessons from disease. Curr Opin Genet Dev. 2009;19:220-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The ciliopathies: An emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125-48.
- [CrossRef] [PubMed] [Google Scholar]
- Renal disease in arima syndrome is nephronophthisis as in other joubert-related cerebello-oculo-renal syndromes. Am J Med Genet A. 2004;131:71-6.
- [CrossRef] [PubMed] [Google Scholar]
- The meckel syndrome: Clinicopathological findings in 67 patients. Am J Med Genet. 1984;18:671-89.
- [CrossRef] [PubMed] [Google Scholar]




