

Translate this page into:
Clinical profile and outcome of renal tubular disorders in children: A single center experience
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Tubular disorders form a significant proportion of pediatric kidney diseases and are an important differential diagnosis of failure to thrive (FTT) in children. Data regarding their outcome is scarce from India. We evaluated the clinical profile of these children and studied the outcome in terms of their growth and renal failure. This is a retrospective longitudinal study of all children with renal tubular disorders attending a tertiary care pediatric nephrology center from 2005 to 2010. Growth and renal outcomes were assessed by Z scores and estimated glomerular filtration rate at diagnosis and. The common disorders encountered were distal renal tubular acidosis (d-RTA) (44%), Bartter-like (Bartter's and Gitelman) syndromes (22%) followed by hereditary Fanconi syndrome (cystinosis and idiopathic Fanconi syndrome) (13%) and few cases of nephrogenic diabetes insipidus, hypophosphatemic rickets and idiopathic hypercalciuria. Male: female ratio was 1.22. The median age at diagnosis was 1.5 (range 0.13-11) years. Growth failure was the presenting feature in 86% of children followed by polyuria (60%) and bone deformities (47%). In 60% of children with hereditary Fanconi syndrome, nephropathic cystinosis was diagnosed, all of whom progressed to stage III chronic kidney disease (CKD) within 3.41 ± 1.42 years. With appropriate therapy, catch-up growth was noted in d-RTA and Bartter syndrome. Renal tubular disorders usually present with FTT. d-RTA is the most common etiology followed by Bartter-like syndrome. Renal function is preserved in all these disorders except for nephropathic cystinosis, who ultimately progressed to CKD. With appropriate and inexpensive therapy, these children do grow well.
Keywords
Chronic kidney disease
distal renal tubular acidosis
estimated glomerular filtration rate
nephrogenic diabetes insipidus

Introduction
Renal tubules play an important role in fluid, electrolyte, and acid-base homeostasis. Defect of these functions can give rise to a host of disorders.[12] These disorders can be inherited or acquired. In India, the data is especially scarce with regard to prevalence and clinical course of these disorders. Early diagnosis and prompt therapeutic interventions can improve the overall clinical outcome of these children. Moreover, with improved management strategies, many of these children are reaching adulthood. Therefore knowledge about their natural history and long-term outcome is of particular importance. We describe the clinical profile of various tubular disorders followed-up in our center ans outcome in terms of growth and progression to renal failure.
Subjects and Methods
A retrospective longitudinal study of all children with renal tubular disorders attending our center for 5 years (2005-2010) was done. Children with tubular disorders secondary to drugs and transient in nature were excluded. Furthermore, children with no follow-up in last 5 years were excluded.
Growth assessment was done by serial record of height and weight. The height standards of the Indian academy of pediatrics growth charts were used to calculate age-related standard deviation scores (SDS) for height.[3] Growth was analyzed as Z scores for the age and sex and compared.
The glomerular function was assessed by estimated glomerular filtration rate (eGFR) calculated using Schwartz formula.[4] Chronic kidney disease (CKD) was defined in the presence of a permanent reduction in GFR (e-GFR; <60 ml/min/1.73 m2 Kidney Disease Outcomes Quality Initiative < stage 3).[5] The term CKD is used here independent of the degree of tubular impairment.
Numerical data was expressed as proportion, median with range, and mean with 95% confidence interval as applicable. Significance was tested with appropriate tests like Student's t-test and a P < 0.5 was taken as significant. OpenEpi, Open Source Epidemiologic Statistics for Public Health, Version 2.3.1. www.OpenEpi.com, updated 2013/04/06, was used for analysis.
Results
We screened the case records of 85 children with renal tubular disorders of which 67 were found to be eligible and regularly followed at the pediatric nephrology outpatient clinics of our hospital during the study period. Duration of follow-up in our clinic was 0.08-14 years (median 3.25 years).
Spectrum of renal tubular disorders
A wide range of renal tubular disorders were seen. Distal renal tubular acidosis (d-RTA), hereditary Fanconi (including cystinosis) and Bartter-like syndromes were common causes encountered. The detailed description of the spectrum with other demographic details is summarized in Table 1.

Age at diagnosis
The median age at diagnosis of all renal tubular disorders was 1.5 years (range 0.13-11 years). The age at the time of diagnosis was below 1.0 years in 40%, 1-3 years in 21%, 3-6 years in 17%, 6-10 years in 7%, and above 10 years in 2% of all children. Initial clinical manifestations often appeared some months or even years before a definite diagnosis of renal tubular disorder was made.
Clinical presentation
Most frequently encountered initial symptoms were failure to thrive (FTT) (88%), followed by polyuria (>4 ml/kg) and bone deformities. Photophobia was observed in five of the six children of cystinosis; no other children had photophobia. Sudden onset of weakness, due to hypokalemia was seen in five children (one in Bartter and four in d-RTA). In idiopathic hypercalciuria, isolated gross or microscopic hematuria led to the diagnosis. The symptomatology of the various tubular disorders is summarized in Table 2.

Fifty-two percent of children had evidence of rickets either clinically or radiological or both. One or more osteotomies were performed in two children (mainly in hypophosphatemic rickets) before diagnosis was established and concerned exclusively the tibia. On slit lamp examination, cystine crystals were seen in all the six children with cystinosis.
History of polyhydramnios was seen in 70% of children with Bartter syndrome and was limited to this condition only. One child with d-RTA had h/o oligohydramnios. Nine children were born preterm, seen in Bartter (28%) and d-RTA (13%) group only. Twenty-eight percent of Bartter children and 13% of d-RTA group had documented low birth weight, suggesting severity of the salt losing defect from intrauterine life. Consanguinity was seen in 13/67 children (20%) only.
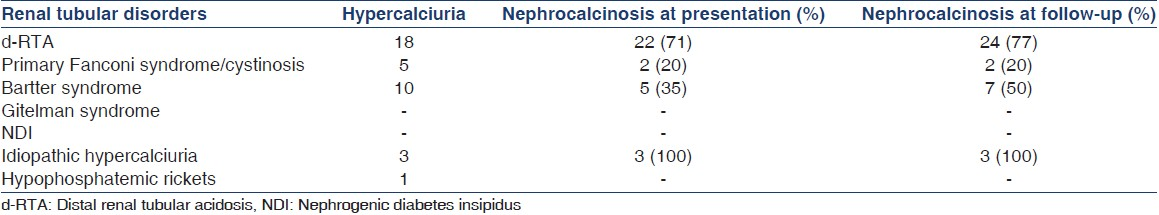
Table 3 shows details of nephrocalcinosis in the cases. Appropriate treatment was instituted and regular follow-up emphasized. All children with cystinosis received standard CKD care. Cysteamine therapy was not available in India at that time, hence not given.
Growth outcome
Height for age
With appropriate treatment and on follow-up, it was seen that significant improvement in height was noticed in d-RTA only (P < 0.001). There was no significant catch-up growth in children with Fanconi syndrome. Though, there was a trend toward catch-up growth in the rest all children, this did not translate into statistical significance. Height for age Z scores are shown in Figure 1.
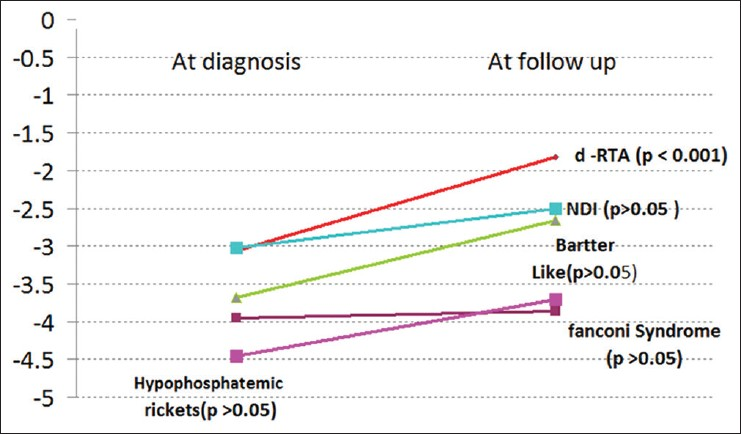
- Height for age – Z scores, with appropriate therapy improvement of height was seen in all, but was statistically significant in distal renal tubular acidosis only (except cystinosis, where no catch-up growth was seen)
Weight for age
Significant improvement in weight of these children was noticed in d-RTA and Bartter syndrome cohort. Children with cystinosis did not grow well due to onset of CKD. Weight for age – Z scores are shown in Figure 2.
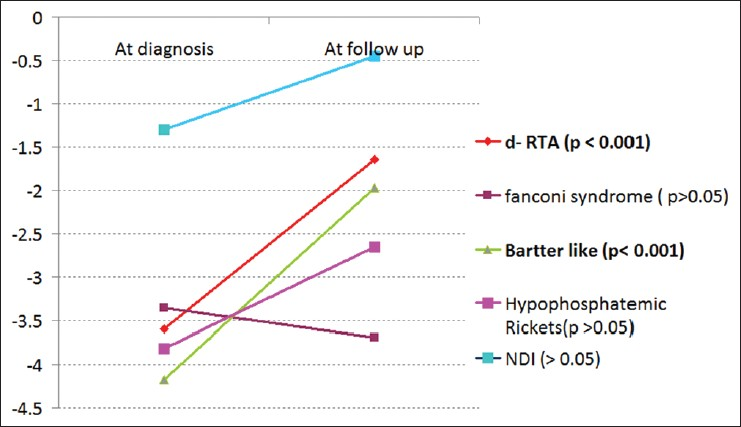
- Weight for age – Z scores, statistical significant weight gain is seen in distal renal tubular acidosis and Bartters syndrome, nephrogenic diabetes insipidus and hypophosphatemic rickets showed trend of improvement. While children with Fanconi syndrome (cystinosis) had no improvement
Renal function outcome
Progression to CKD was observed in all children with cystinosis the time for progression was 3.41 ± 1.42 years. No child required renal replacement therapy yet. One child with Bartter syndrome had AKI at presentation due to dehydration episode, which improved completely.
Discussion
The renal tubular disorders seen at our center represents a wide spectrum, but lacks some rare disorders reported in the literature.[12] In our experience, d-RTA is the most common renal tubular disorder comprising 46% of all tubular disorders, followed by Bartter - like syndrome, nephropathic cystinosis presenting as Fanconi syndrome and hypophosphatemic rickets. This is in stark contrast to the western world where nephropathic cystinosis, hypophosphatemic rickets, and idiopathic hypercalciuria are among the leading causes.[2] Other rare causes of hereditary Fanconi syndrome like galactosemia and Wilson's disease were not seen in our series. Cystinosis is the most common cause of Fanconi syndrome in our series consistant with published literature.[2] Regarding Idiopathic hypercalciuria the incidence may be reflecting the “proverbial tip of iceberg” and is underestimated in this series because most of these children do not get referred to a pediatric nephrology center.
Age of presentation ranged 1.5 months to 11 years with a median of 1.5 years. As expected, in patients with a more severe symptomatology (e.g. primary Fanconi syndrome, d-RTA, neonatal Bartter syndrome), the diagnosis was made earlier than in those with a more silent course in whom it was often delayed until later childhood (Gitelman syndrome, hypophosphatemic rickets, idiopathic hypercalciuria). The age at presentation of various diseases are comparable with those published in the literature.[6789] However, it was interesting to note that there was a significant gap between the onset of symptoms and diagnosis in some children. This could be because of a missed diagnosis by the physician at the first presentation or delay in seeking medical attention, which not uncommon in developing countries. In the series of d-RTA by Bajpai et al., the onset of symptoms was at the age of 1.8 years (3 months to 7.5 years) and at diagnosis 6 years (1.5-13 years).[6] An early diagnosis may be associated with early initiation of therapy and thus a better outcome. Increasing awareness of primary care physicians/pediatricians about tubular disorders may ensure early referral and diagnosis.
Tubular dysfunction usually was first manifested by polydipsia/polyuria and urinary loss of electrolytes and bicarbonate. In some disorders, (idiopathic hypercalciuria) microscopic hematuria on routine urine examination gave a clue to the diagnosis of the disease. Most frequently encountered initial symptoms were FTT, followed by polyuria and bone deformities. The main bone changes observed were rickets. Bone deformities of variable degree were found in 52% of all patients. Therefore, cases of refractory rickets should be considered for diagnosis of renal tubular disorder. It was unfortunate that one or more osteotomies of the tibia were performed in two children with hypophosphatemic rickets. Doing such surgeries to correct “bent bone” will fail until and unless the disease is diagnosed and managed. Recurrent fevers with dehydration requiring intravenous fluids were seen in a quarter to third of children with polyuric disorders and can be an important clinical diagnostic clue. Other features, which could be found are recurrent episodes of tetany, salt craving. Hypokalemia and acid base abnormalities were important presenting feature. Hypokalemia can present as subclinical or as weakness or quadriparesis. Acidosis can present as quite tachypnea. Photophobia, secondary to cystine crystals in the cornea is pathognomonic for cystinosis[10] and was seen in all the six children with Fanconi syndrome. This can be used to clinch the diagnosis in resource limited setting where cystine levels are either not available or not affordable as all children have corneal crystals by 16 months.[11] Gitelman syndrome and idiopathic hypercalciuria children did not show these features. These have presenting features consistent with published literature.[16789] The incidence of sensory neural hearing loss seems to be less than that published.[1112] This could be because of underestimation of the true incidence because all children with d-RTA did not undergo a formal hearing assessment. Our study also reconfirms that antenatal history is very important in suspicion and clinical diagnosis of tubular disorders. There could be history of polyhydramnios, prematurity and low birth weight.
Nephrocalcinosis is an important presenting feature as well as an important outcome variable in tubular disorders.[13] Nephrocalcinosis was observed in 32 patients (47%) at presentation. In our series, d-RTA is the most common cause of nephrocalcinosis affecting more than half of children (with d-RTA). Other important causes were idiopathic hypercalciuria, Bartter syndrome, and primary Fanconi syndrome.
On follow-up, two more children with d-RTA and one more with Bartter syndrome developed nephrocalcinosis. There was no resolution of nephrocalcinosis on follow-up. The high rate of nephrocalcinosis at presentation may be because of delay in diagnosis and institution of therapy. This problem may be common to all developing nations and has been evident from other centers from India.[613] The authors speculate that more awareness among physicians about these disorders may decrease the age at diagnosis and therefore, lessen the proportion of children with nephrocalcinosis at onset.
Growth retardation has been a recognized problem in tubular disorders. However, with adequate treatment, good catch-up growth has been demonstrated.[671214] The cause of growth retardation could include chronic hypokalemia and metabolic acidosis, rickets, nephrocalcinosis, and CKD.[121315] In our series, with appropriate therapy, though only children with d-RTA showed statistically significant catch-up growth, there was the trend toward improvement in growth standard deviation in all except cystinosis children.
In the western world, cystinosis is the most common cause for renal failure in tubular disorders.[216] Our experience was also the same. Cystinosis children were the only group developing progressive CKD. The lower age of developing CKD is a reflection of nonavailablity of cysteamine in Indiaat the time of study, although it can be made available now with import permit. Other causes of CKD in tubular disorders include nonsteroidal antiinflammatory drug toxicity, nephrocalcinosis, and hypokalemic nephropathy.[113] However, such cases were not encountered in our series.
Conclusion
Renal tubular disorders usually present with growth failure. d-RTA is the most common etiology followed by Bartter-like syndrome in this series. Renal function is preserved in all these disorders except for nephropathic cystinosis, who ultimately progressed to CKD. With appropriate and inexpensive therapy, these children do grow well.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Tubular disorders of electrolyte regulation. In: Avner ED, Harmon WE, Niaudet P, eds. Pediatric Nephrology. Baltimore: Lippincott Williams and Wilkins; 2004. p. :729-56.
- [Google Scholar]
- Long-term outcome of paediatric patients with hereditary tubular disorders. Nephron. 1999;83:250-60.
- [Google Scholar]
- IAP growth monitoring guidelines for children from birth to 18 years. Indian Pediatr. 2007;44:187-97.
- [Google Scholar]
- The use of plasma creatinine concentration for estimating glomerular filtration rate in infants, children, and adolescents. Pediatr Clin North Am. 1987;34:571-90.
- [Google Scholar]
- K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am J Kidney Dis. 2002;39(Suppl 1):S1-266.
- [Google Scholar]
- Long-term outcome in children with primary distal renal tubular acidosis. Indian Pediatr. 2005;42:321-8.
- [Google Scholar]
- Clinical outcome of children with primary distal renal tubular acidosis. J Med Assoc Thai. 2011;94:1205-11.
- [Google Scholar]
- Childhood Bartter's syndrome: An Indian case series. Indian J Nephrol. 2010;20:207-10.
- [Google Scholar]
- Bartter's syndrome in Arabic children: Review of 13 cases. Pediatr Int. 1999;41:299-303.
- [Google Scholar]
- Corneal crystals in nephropathic cystinosis: Natural history and treatment with cysteamine eyedrops. Mol Genet Metab. 2000;71:100-20.
- [Google Scholar]
- Distal renal tubular acidosis and its relationship with hearing loss in children: Preliminary report. Iran J Kidney Dis. 2010;4:202-6.
- [Google Scholar]
- Primary distal tubular acidosis in childhood: Clinical study and long-term follow-up of 28 patients. J Pediatr. 1992;121:233-41.
- [Google Scholar]
- Etiology of nephrocalcinosis in northern Indian children. Pediatr Nephrol. 2007;22:829-33.
- [Google Scholar]
- Long-term follow-up of patients with Bartter syndrome type I and II. Nephrol Dial Transplant. 2010;25:2976-81.
- [Google Scholar]
- Longitudinal growth in chronic hypokalemic disorders. Pediatr Nephrol. 2010;25:733-7.
- [Google Scholar]
- Report on management of renal failure in children in Europe, XXIII, 1992. Nephrol Dial Transplant. 1994;9(Suppl 1):26-40.
- [Google Scholar]




