

Translate this page into:
Immunoglobulin A dominant membranoproliferative glomerulonephritis in an elderly man: A case report and review of the literature
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Immunoglobulin A (IgA) dominant membranoproliferative glomerulonephritis (MPGN) is rare, described only as case reports. We report a rare case of an elderly man presenting with rapidly progressive renal failure and nephrotic range proteinuria with histological, immunofluorescence, and ultrastructural findings supporting a diagnosis of IgA dominant MPGN. Autoimmune disease, cryoglobulinemia and infection-associated glomerulonephritis were excluded. Remission was achieved within 3 months of treatment. This case highlights an uncommon diagnosis with a good response to therapy. The differential diagnosis of IgA nephropathy with MPGN-like pattern is discussed.
Keywords
Adult nephrotic syndrome
immunoglobulin A nephropathy
membranoproliferative glomerulonephritis
rapidly progressive renal failure

Introduction
Membranoproliferative glomerulonephritis (MPGN) is sub-grouped according to the findings on immunofluorescence into immunoglobulin (Ig)-positive and negative MPGN. However, Ig-positive IgA dominant MPGN is uncommon, except for case reports in children and in alcoholic cirrhosis.[123] As compared to IgA dominant MPGN, IgA dominant post-infectious glomerulonephritis is now well-recognized and is usually associated with Staphylococcal infections.[45] We report an unusual case of an elderly patient presenting with rapidly progressive renal failure and nephrotic range proteinuria, which on renal biopsy showed a diffuse MPGN-pattern with presence of hyaline thrombi. Immunofluorescence revealed co-dominant IgA and C3 deposits and ultrastructural examination supported the diagnosis of Ig-mediated IgA dominant MPGN.
Case Report
A 55-year-old man presented with edema for 1 month and decreased urine output, fever with chills, breathlessness, and anorexia, nausea and recurrent vomiting for 2 weeks. There was no history of hematuria, cough, sore throat, skin rashes or joint pains. On examination, his blood pressure was elevated (180/90 mmHg). There was no organomegaly. Hematological investigations revealed low hemoglobin (8.8 g/dl), normal total leucocyte (9,300/dl) and platelet (1,20,000/dl) counts with no evidence of hemolysis. Peripheral blood smear for malarial parasite was negative. Biochemical investigation showed elevated serum creatinine (7 mg/dl) and marginally low serum protein (6.9 g/dl) and albumin (3.4 g/dl) levels. On urine examination, the output was 1800 ml, with 4+ proteinuria and active sediments (15–20 red blood cells/hpf, and 12–15 white blood cells/hpf). The 24-h proteinuria was 4.2 g. Serum complement C3 level was 68.8 mg/dl (normal range 60–120 mg/dl) and C4 was 17.6 mg/dl (normal range 15–25 mg/dl). Serology for anti-nuclear, anti-glomerular basement membrane (GBM) and anti-neutrophil cytoplasmic antibodies was negative. He was HIV, HBsAg and hepatitis C virus negative. Serum cryoglobulins were also negative. A clinical diagnosis of rapidly progressive renal failure and nephrotic range proteinuria, with a possibility of crescentic glomerulonephritis was made and a renal biopsy performed after three sessions of hemodialysis.
Light microscopy showed 22 enlarged glomeruli with lobular accentuation, thickening of glomerular capillary walls, endocapillary proliferation, segmental neutrophilic exudate, basement membrane thickening and an occasional focus of loop necrosis. Hyaline thrombi were also seen [Figure 1a]. Silver stain showed diffuse splitting of basement membrane [Figure 1a, inset]. No crescents were found. Tubules showed features of acute tubular necrosis. The interstitium and blood vessels were unremarkable.
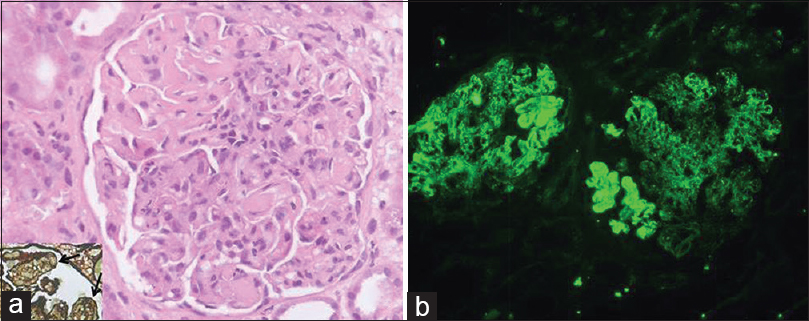
- Renal histology showing enlarged glomerulus with endocapillary proliferation, basement membrane thickening, hyaline thrombi (a) and widespread splitting of basement membrane on silver stain (arrows; inset); immunofluorescence showed strong (4+) capillary wall and mesangial immune deposits of immunoglobulin A (b) (H and E, ×400; FITC, ×200)
On immunofluorescence, glomeruli showed co-dominant 4+ granular capillary wall and few mesangial deposits IgA and C3 [Figure 1b]. IgG was 2+ with a similar pattern. IgM and C1q were negative. No monoclonal deposits were found; kappa and lambda being of similar intensity.
Ultrastructural examination showed extensive subendothelial electron dense deposits with GBM duplication and mesangial interposition [Figure 2]. Capillary luminal and few mesangial deposits were also present. No subepithelial deposits were found. There was focal foot process effacement.
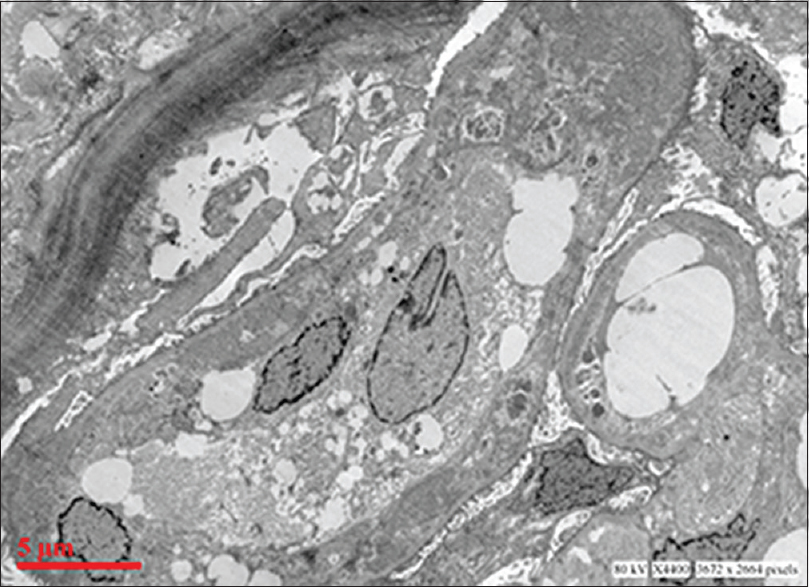
- Electron micrograph showing extensive glomerular subendothelial electron dense deposits, duplication of basement membrane and mesangial interposition (uranyl acetate and lead citrate)
Considering the histology, pattern and type of immune deposits and ultrastructural features, a diagnosis of IgA-dominant Ig mediated MPGN was considered. IgA Nephropathy with MPGN-like pattern (M1E1S0T0 [Oxford classification]; HAAS Class IV) could be another possibility; however, a diagnosis of IgA-dominant MPGN was more likely in view of the morphological and ultrastructural features and is discussed.
The blood culture was sterile, and urine culture showed growth of Enterococcus faecalis. The patient was administered antibiotics and intravenous steroids followed by oral steroids and cyclophosphamide therapy. At discharge, the serum creatinine was 4.2 mg%. On follow-up, remission was achieved within 3 months. Steroid was tapered, and a maintenance therapy of Azathioprine and angiotensin receptor blockers was started. On last follow-up, 8-month after presentation, the serum creatinine was 1.2 mg% with no proteinuria and bland urinary sediments.
Discussion
Our case presented with rapidly progressive renal failure and nephrotic range proteinuria. There was no clinical or serological evidence of systemic illness. The only evidence of infection was positive urine culture, which could be the reason for the appearance of fever with chills 2 weeks after the onset of edema. Urinary tract infection is a secondary event rather than the etiological factor for the renal disease. Based on histology, immunofluorescence and ultrastructural features a diagnosis of IgA-dominant MPGN was made. The patient responded to treatment and remission was achieved within 3 months.
Membranoproliferative glomerulonephritis is a pattern of glomerular injury easily recognized by light microscopy that was historically subtyped into three categories (MPGN type I–III) on the basis of histology and electron microscopy. However, recent classification of MPGN is based primarily on the differences in immunofluorescence, and subtypes MPGN as Ig-positive and negative that is, C3 glomerulopathy.[6]
In our patient, renal biopsy on light microscopy showed MPGN pattern, Ig deposits were present on immunofluorescence and on electron microscopy subendothelial dense deposits were found, therefore, the diagnosis is Ig-mediated MPGN with dominant IgA deposits. However, there are some conflicting features. Ig mediated MPGN is associated with activation of classical complement pathway leading to low or normal C3 and low C4. In this patient, hypocomplementenemia was not present, though both serum C3 and C4 levels were towards the lower limit of normal. The other feature which is unusual for MPGN is the rapid response to therapy. MPGN in adults shows progression to end-stage renal disease in 40% patients within 10-years of diagnosis.[7]
The other differential diagnosis is primary IgA nephropathy Primary IgA nephropathy is a histologically heterogeneous disease, which can show minimal glomerular lesions to focal or diffuse, segmental or global mesangial hypercellularity and/or endocapillary proliferation, however, diffuse MPGN-like pattern is not described. Secondly, presence of extensive subendothelial deposits without prominent mesangial or paramesangial deposits is not seen in IgA nephropathy. In a study of 244 patients of IgA nephropathy, Haas M found no case with MPGN-like histology. He described small peripheral capillary wall (subepithelial and/or subendothelial) deposits in 23% of cases.[8]
Immunoglobulin A dominant post-infectious glomerulonephritis is now a well-recognized entity, particularly seen in the elderly.[45910] The majority of cases are associated with Staphylococcal infections.[9] It presents clinically as proteinuria (100%), acute kidney injury (98%) and hematuria (95%).[10] IgA dominant post-infectious glomerulonephritis shows variable histological features ranging from mesangial hypercellularity to diffuse proliferative glomerulonephritis and dominant or co-dominant IgA deposits by immunofluorescence. Electron microscopy is essential for differentiating IgA nephropathy from cases of IgA dominant post-infectious glomerulonephritis that are in the resolving or persistent phase. Electron microscopy resembles post-streptococcal glomerulonephritis and typically shows subepithelial “hump”-shaped electron-dense deposits.
Renal disease resembling IgA dominant MPGN has been described previously in children and in alcoholic liver disease.[123] Iitaka et al., reported two pediatric patients of nephrotic syndrome showing histological changes compatible with type-I MPGN with presence of strong co-dominant IgA and C3 immune deposits on immunofluorescence along glomerular capillary walls. Both patients responded to treatment in a mean duration of 4 years.[1] A case of an infant with incidentally detected mild proteinuria and microscopic hematuria, which showed type-I MPGN-like lesion on histology and co-dominant IgA, IgG and C3 deposits on immunofluorescence, has been described. The urinary protein disappeared within 6-month of steroid therapy with complete remission achieved within 8-months.[2] MPGN-like pattern with dominant IgA deposits has also been reported in a patient with alcoholic cirrhosis.[3]
Thus, we report an unusual patient with IgA dominant MPGN, which is a rare disease not associated with systemic illness or infection. This case illustrates the importance of timely initiation of treatment before the onset of histological sclerotic lesions in MPGN.
Source of Support: Nil
Conflict of Interest: None declared.
References
- IgA-associated glomerulonephritis with membranoproliferative glomerulonephritis-like pattern in two children. Clin Exp Nephrol. 2003;7:284-9.
- [Google Scholar]
- Infantile immunoglobulin A nephropathy showing features of membranoproliferative glomerulonephritis. Tohoku J Exp Med. 2012;228:253-8.
- [Google Scholar]
- Membranoproliferative glomerulonephritis with IgA deposits in patients with alcoholic cirrhosis. Pathologica. 1986;78:469-78.
- [Google Scholar]
- Staphylococcus infection-associated glomerulonephritis mimicking IgA nephropathy. Clin J Am Soc Nephrol. 2006;1:1179-86.
- [Google Scholar]
- Immunoglobulin A-dominant postinfectious glomerulonephritis: Frequent occurrence in nondiabetic patients with Staphylococcus aureus infection. Hum Pathol. 2011;42:279-84.
- [Google Scholar]
- Membranoproliferative glomerulonephritis and C3 glomerulopathy: Resolving the confusion. Kidney Int. 2012;81:434-41.
- [Google Scholar]
- Histologic subclassification of IgA nephropathy: A clinicopathologic study of 244 cases. Am J Kidney Dis. 1997;29:829-42.
- [Google Scholar]
- IgA-dominant postinfectious glomerulonephritis: A report of 13 cases with common ultrastructural features. Hum Pathol. 2008;39:1309-16.
- [Google Scholar]
- IgA dominant post-infectious glomerulonephritis: Pathology and insights into disease mechanisms. Diagn Histopathol. 2013;19:175-81.
- [Google Scholar]




