

Translate this page into:
Primary Hyperoxaluria Type 1 with Homozygosity for a Double-mutated AGXT Allele in a 2-year-old Child
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Primary hyperoxaluria (PH) Type 1 is a rare, genetic disorder caused by deficiency of the liver enzyme alanine-glyoxylate aminotransferase, which is encoded by AGXT gene. We report a 2-year-old South Indian Tamil child with nephrocalcinosis due to PH Type 1, in whom a homozygous genotype for two missense mutations in the AGXT gene was found: first, a C to G transversion (c. 32C>G) in exon 1 resulting in the amino acid substitution p.Pro11Arg; second, a T to A transversion (c. 167T>A) in exon 2 resulting in p.Ile56Asn. A therapy based on potassium citrate and pyridoxine was started. This is the first report of molecular testing-proven childhood onset-PH Type 1 from South India and is notable for the co-occurrence of two missense mutations in one AGXT allele, which might lead to different and more severe phenotype than each mutation alone. To the best of our knowledge, AGXT allele carrying two already known mutations has not been previously reported.
Keywords
AGXT gene
double-mutated allele
missense mutation
primary hyperoxaluria Type 1

Introduction
Primary hyperoxaluria (PH) is a rare, autosomal recessive disorder that commonly presents in childhood with nephrocalcinosis and/or nephrolithiasis and progresses to chronic renal failure. Three genes are known to be responsible for the disease: alanine-glyoxylate aminotransferase (AGXT, PH Type 1), glyoxylate reductase/hydroxypyruvate reductase (GRHPR, PH Type 2), and 4-hydroxy-2-oxoglutarate aldolase (HOGA1, PH Type 3). PH Type 1 is the most frequent and severe form.[1] The AGXT gene coding the AGT enzyme is located at 2q37.3 and consists of 11 exons. We herein describe a child who was diagnosed to have two homozygous missense mutations in the AGXT gene.
Case Report
A 2-year-old male child from Villupuram, Tamil Nadu, was referred to us for a clinical suspicion of renal stones. The child was developmentally normal for age. Poor weight gain was reported by the parents, while there was no history of diarrhea, polyuria, polydipsia, pallor, dysuria, recurrent respiratory tract infections, or anorexia. He was born to third-degree consanguineous Tamil parents. At birth, he weighed 3.5 kg and his perinatal history was uneventful. He was immunized according to the age. At the age of 24 months, he weighed 9.7 kg (−1–−2 standard deviation [SD]), height 71 cm (−2–−3 SD), head circumference 46 cm, and vital signs and blood pressure (93/62 mmHg) being normal. No pallor, icterus, clubbing, edema, rickets, or lymphadenopathy was noted. Cardiovascular, abdominal, central nervous system and chest examinations were normal. No renal masses were appreciable. The abdominal X-ray showed nephrocalcinosis [Figure 1].

- Abdominal X-ray of the child showing nephrocalcinosis
He was fourth in birth order. His elder sister died at 9 years of age as a result of end-stage renal disease due to nephrocalcinosis. She had persistent vomiting at 3 months of age. She was admitted to another hospital where renal stones and high urinary oxalate levels (80 mg/24 h) were detected. A therapy based on potassium citrate and pyridoxine was suggested (at the age of 4 months), but was administered to the child only for 1 year, and then discontinued by the parents. At 8 years of age, she was found to have urinary tract infection and high serum creatinine levels (8.2 mg/dl). She was then kept on biweekly dialysis for 3 months and died at 9 years of age at home. No genetic testing was performed.
The two other siblings, aged 6 and 4 years, were healthy and did not have nephrocalcinosis, deranged renal function, or high urinary oxalate levels. Their parents were healthy; no further cases of nephrolithiasis were reported in the family.
In view of the family history and presence of nephrocalcinosis on abdominal X-ray, the child was investigated further: 24 h urinary oxalate level was high (75.7 mg/24 h), blood urea 32 mg/dl, serum creatinine 0.4 mg/dl (estimated glomerular filtration rate 80 mL/min/1.73 m2), serum calcium 9.2 mg/dl, phosphate 4.0 mg/dl, sodium 136 mEq/L, potassium 3.8 mEq/L, and hemoglobin 9.7 g/dl. The spot urine calcium: Creatinine ratio was 0.1 (normal for age). Venous blood gas analysis showed serum bicarbonate level 22 mEq/L. Renal ultrasonography showed medullary nephrocalcinosis.
Molecular analysis of the AGXT gene
The entire coding region of the AGXT gene (RefSeq NM_000030 2, Gene ID: 189) was amplified by polymerase chain reaction from genomic DNA extracted from blood. Analysis of the AGXT gene by means of Sanger sequencing[2] revealed the presence of two single nucleotide substitutions in homozygous state: a transversion from C to G (c. 32C>G) in exon 1 resulting in the missense substitution p. Pro11Arg and a transversion from T to A (c. 167T>A) in exon 2 resulting in the missense substitution p. Ile56Asn [Figure 2]. Both variants have previously been reported and are classified as pathogenic. The other polymorphic sites of the gene were found in homozygosis and corresponded to the major haplotype.[3] Following the current nomenclature,[4] the AGXT gene mutations detected in this patient are described as: c.[32C>G; 167T>A];[32C>G; 167T>A] at the DNA level and p.[Pro11Arg; Ile56Asn];[Pro11Arg; Ile56Asn] at the protein level. Thus, the diagnosis of PH Type 1 was confirmed. Genetic testing of the parents was advised but not performed since they did not agree to undergo any further analysis.
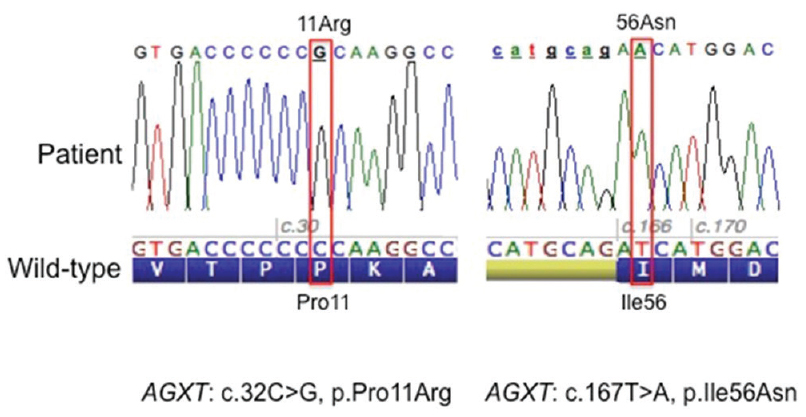
- Sequences and electropherograms showing the two missense mutations identified in the affected child; on the left sequence around codon 11 and the right sequence around codon 56. The wild-type nucleotide and protein sequence (amino acid in one letter code) are reported below
The patient was treated with potassium citrate (0.5 mEq/kg/day) along with pyridoxine (5 mg/kg/day). He was advised to increase fluid intake to 3 L/m2/day, avoid oxalate-rich foods (such as tomato, spinach, and beetroot,), and take a low salt diet.
Discussion
PH Type 1 (OMIM 259900) is an autosomal recessive disorder of glyoxylate metabolism resulting from a functional defect in the AGXT-peroxisomal liver-specific enzyme.[3] The phenotype expression depends on the increased excretion of calcium oxalate with consequent nephrolithiasis and nephrocalcinosis as main symptoms and eventually systemic oxalosis.[156] The disease is characterized by clinical and allelic heterogeneity, with a wide range of signs and symptoms and a variable degree of severity,[7] to some extent related to the genes causing mutations.[5689] An early diagnosis is important to prevent or slow down the end-stage renal disease.
The age at onset is variable: 15% of children become symptomatic before the age of 1 year and 50% before the age of 5 years.[6] The severe infantile form is associated with failure to thrive, nephrocalcinosis (in 91%), nephrolithiasis, and/or bladder/urethral stones with hematuria, dysuria, urinary tract infections, and renal colic. Early end-stage renal failure eventually sets in. During childhood or adolescence, the disorder is characterized by recurrent urolithiasis and progressive renal failure. Some individuals may be asymptomatic until adulthood with occasional or recurrent renal stones, sometimes resulting in obstructive features. In our patient, the main symptom at onset was the failure to thrive and hyperoxaluria testing was performed after the death of his affected sister.
Although the occurrence of nephrocalcinosis and nephrolithiasis is known in Indian children,[10] screening for hyperoxaluria is rarely performed.[11] The diagnosis of PH Type 1 is suspected in any child with an elevated urinary oxalate excretion (>0.5 mmol [45 mg]/1.73 m2/day).[1] PH Type 1 can be confirmed by identification of biallelic pathogenic variants in the AGXT gene. Reduced AGXT enzyme activity in liver biopsy specimens, once considered as the gold standard, has now been largely replaced by molecular genetic testing.[1512]
More than 200 AGXT mutations have been described,[13] spanning the entire gene. The three most common mutations, c. 508G>A (p. Gly170Arg), c. 33dupC (p. Lys12Glnfs*156), and c. 731T>C (p. Ile244Thr), account for approximately 30%, 11%, and 6% of AGXT mutant alleles, respectively.[1214] In populations with high level of consanguinity as seen in and around Tamil Nadu in South India, the frequency of autosomal diseases is higher and even rare mutations may become evident. This is exemplified in our case, where two separate mutations, c. 32C>G (p. Pro11Arg) and c. 167T>A (p. Ile56Asn) with an allele frequency of 0.0001264% and 0.0003028% respectively in South Asian population as reported in ExAC database, presented together in homozygous state. Hence in any case suspected with PH1, sequencing of entire AGXT coding region is strongly recommended.[1]
Genetic testing is important for an early and definitive diagnosis of PH Type 1, considering the high incidence of early progression to renal insufficiency. The identification of the causative mutations permits to start an optimal therapy, such as pyridoxine therapy in those patients who are carriers of misfolding mutations (p. Gly170Arg and p. Phe152Ile).[1] A correct diagnosis is most important to identify the need for liver–kidney transplantation.[15] Genetic testing also enables prenatal counseling and prenatal diagnosis if the proband mutations are known.
Interestingly, one of our patient's AGXT mutations causes the p. Pro11Arg substitution. Proline 11 indeed is known to be involved in the intracellular AGT targeting to peroxisomes; any mutation of this codon could implicate AGT mistargeting with a consequently reduced enzymatic activity, in general, without being per se pathogenic. However, the particular c. 32C>G, p. Pro11Arg variant is known to be pathogenic on the basis of in vitro studies, showing the reduction to <2% of normal enzymatic activity.[16] The other mutation c. 167T>A, p. Ile56Asn is known to produce an unstable enzyme.[17] This is the first case in which these two mutations are found on the same allele and might be leading to a severe deficiency of enzyme activity, with an early onset of disease.
In literature, few Indian cases of PH Type 1 have been reported. Chanchlani et al.[11] and Sethi et al.[18] described North Indian children affected by PH Type 1 who were homozygous for the c. 302T>C (p. Leu101Pro) AGXT mutation. The two South Indian patients described by Dutta et al.[19] were homozygous for another mutation in exon 4 (c. 447_454delGCTGCTGT, p. Leu151Asnfs*14); their clinical features comprise a late-onset disease in adulthood.
Several authors described AGXT alleles carrying two variants, of which one is known to be pathogenic and the other therefore remaining of uncertain pathogenic significance.[16] To the best of our knowledge, AGXT allele carrying two already known mutations has not been previously reported.
In PH Type 1, a high fluid intake (at least 3 L/m2/day) is recommended.[1] Vitamin B6 is recommended in any patient with confirmed PH Type 1; starting at a dose of 5 mg/kg/day and not exceeding 20 mg/kg/day, aiming to decrease the urinary oxalate concentration by 30%. Calcium oxalate crystallization inhibition by the use of alkalinization with oral potassium citrate at the rate of 0.3–0.5 mmoL/kg is recommended as long as the glomerular filtration rate is preserved.[1] This may be replaced by sodium bicarbonate appropriate to glomerular filtration rate and plasma potassium. A restriction in oxalate intake is of limited value as the main source of oxalate is endogenous, and intestinal oxalate absorption is lower in PH Type 1 patients compared to normal subjects; however, some experts relying on precautionary principle recommend avoiding oxalate-rich foods (such as spinach, tomato, and beetroot) in the diet.[1] Other dietary measures include a low-salt diet, avoiding excessive Vitamin C and D, and not restricting dietary calcium. Despite the ability of Oxalobacter formigenes to metabolize oxalate, there is as yet no evidence that probiotics can significantly decrease the urinary oxalate concentrations in PH Type 1 patients.[1]
The case described herein is the first report of molecular testing-proven childhood onset-PH Type 1 from South India and is notable for the observed homozygosity of a double-mutated AGXT allele. It is also pertinent to note that these two mutations encountered in our patient, although reported in the AGXT mutation database,[13] are different from those reported in other Indian children or adults.[111819] Considering the scanty literature regarding mutational analysis in childhood-onset PH Type 1 from India, in general, and South India, in particular,[19] this work has the merit to expand the spectrum of genotypes encountered in Indian PH Type 1 patients.
Financial support and sponsorship
The study was supported by RILO 2015 University of Torino (to Daniela Giachino).
Conflicts of interest
There are no conflicts of interest.
References
- Primary hyperoxaluria type 1: Indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012;27:1729-36.
- [Google Scholar]
- Updated genetic testing of Italian patients referred with a clinical diagnosis of primary hyperoxaluria. J Nephrol 2016 Mar 5 [Epub ahead of print]
- [Google Scholar]
- Primary hyperoxaluria. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, eds. The Metabolic and Molecular Bases of Inherited Disease. Vol Vol. II. New York: McGraw-Hill; 2001. p. :3323-67.
- [Google Scholar]
- HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564-9.
- [Google Scholar]
- Primary hyperoxaluria type 1. 1993-2016. GeneReviews®. Seattle (WA): University of Washington, Seattle; URL https://www.ncbi.nlm.nih.gov/books/NBK1283
- [Google Scholar]
- Primary hyperoxaluria: Report of an Italian family with clear sex conditioned penetrance. Urol Res. 2008;36:309-12.
- [Google Scholar]
- Plasma calcium oxalate supersaturation in children with primary hyperoxaluria and end-stage renal failure. Kidney Int. 1999;56:268-74.
- [Google Scholar]
- Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int. 2014;86:1197-204.
- [Google Scholar]
- Etiology of nephrocalcinosis in Northern Indian children. Pediatr Nephrol. 2007;22:829-33.
- [Google Scholar]
- Common mutation underlying primary hyperoxaluria type 1 in three Indian children. Indian J Nephrol. 2012;22:459-61.
- [Google Scholar]
- Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney Int. 2004;66:959-63.
- [Google Scholar]
- 2015. Primary Hyperoxaluria Mutation Database. Available from: https://www.uclh.nhs.uk/OurServices/ServiceA-Z/PATH/PATHBIOMED/BIO/Pages/Phmdatabase.aspx
- Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol. 2015;26:2559-70.
- [Google Scholar]
- A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003;18:986-91.
- [Google Scholar]
- Primary hyperoxaluria type 1: Update and additional mutation analysis of the AGXT gene. Hum Mutat. 2009;30:910-7.
- [Google Scholar]
- Allele-specific characterization of alanine: Glyoxylate aminotransferase variants associated with primary hyperoxaluria. PLoS One. 2014;9:e94338.
- [Google Scholar]
- Primary hyperoxaluria type 1 with a novel mutation. Indian J Pediatr. 2009;76:215-7.
- [Google Scholar]
- Recurrent truncating mutations in alanine-glyoxylate aminotransferase gene in two South Indian families with primary hyperoxaluria type 1 causing late onset end-stage kidney disease. Indian J Nephrol. 2016;26:288-90.
- [Google Scholar]




