

Translate this page into:
A Rare Case Report of Emphysematous Cyst Infection in Autosomal Dominant Polycystic Kidney Disease
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
A 38-year-old non-diabetic female, a known case of autosomal dominant polycystic kidney disease (ADPKD) for the past 1 year, presented to our outpatient clinic with history of fever and right-sided flank pain for 1 month. Family history was significant as her father also had ADPKD and expired at the age of 50 years due to advanced renal failure. Examination revealed bilateral nodular abdominal masses with localized tenderness over the right flank. Investigations revealed neutrophilic leukocytosis with total leukocyte count of 15,000 (80% polymorphs), and urinalysis showed 40–50 pus cells per high-power field with a serum creatinine of 2.7 mg/dL. Blood and urine cultures were sterile. Ultrasonography of the abdomen revealed bilateral enlarged kidneys with multiple cysts. Subsequently, contrast-enhanced computed tomography of the abdomen revealed bilateral polycystic kidneys with gas formation in the right polar cyst [Figure 1]. The patient was empirically started on intravenous antibiotic piperacillin tazobactam on day 1 of hospitalization. Subsequently, she underwent cyst aspiration and percutaneous drainage of the cyst through pigtail catheter. Cyst fluid analysis revealed numerous pus cells and culture grew Escherichia coli sensitive to piperacillin tazobactam. The patient improved symptomatically, and pigtail was removed after 5 days when the drainage subsided. The antibiotic was continued for 6 weeks, and serum creatinine decreased to 1.5 mg/dL after 6 weeks of follow-up.
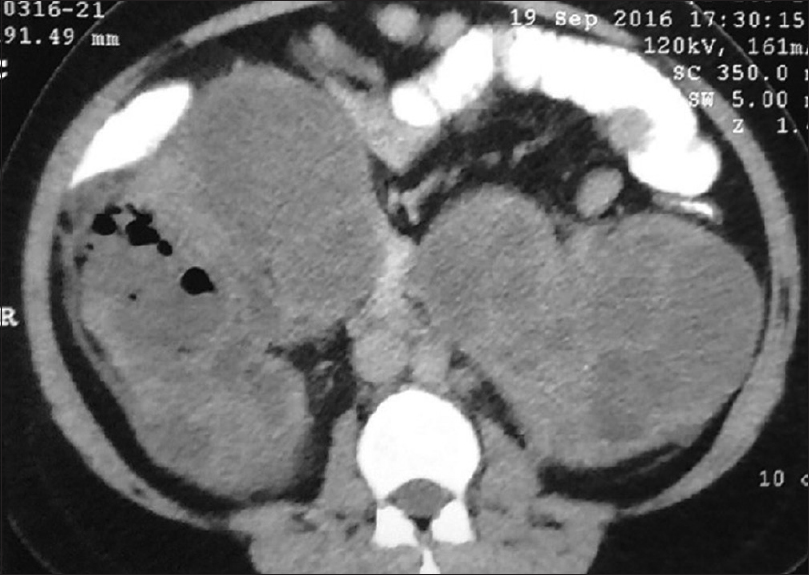
- Contrast enhanced computed tomography of abdomen showing multiple cystic lesions in both the kidneys with air foci in right polar cyst
ADPKD is one of the most common hereditary disorders leading to end-stage renal disease. Approximately 30%–50% of patients with ADPKD have one or more renal infection during their lifetime.[1] However, cyst infections with gas-forming organisms are rare with few case reports in literature. Emphysematous polycystic cyst infection (EPCI) is life-threatening and needs to be differentiated from emphysematous pyelonephritis which is characterized by the presence of gas in the collecting system, perirenal tissues, and absence of gas in the cysts. To date, antibiotic therapy combined with nephrectomy was done for a majority of cases in published literature. Fluoroquinolones are usually the preferred antibiotics of choice because of their lipophilic properties that lead to an increased diffusion into infected cysts and their bactericidal activity against Gram-negative enteric pathogens.[2] Intravenous antibiotic therapy alone may be insufficient to resolve emphysematous cyst infection considering disease severity and poor prognosis. A percutaneous cystostomy drainage and intracystic antibiotic irrigation may be one as the alternative therapeutic options to control emphysematous cyst infection instead of nephrectomy in patients with ADPKD. Despite all measures, mortality in emphysematous cyst infections remains high.
Till now, only seven cases have been described in literature of EPCI and only two of these patients improved with cyst drainage and antibiotics alone without requiring nephrectomy. To conclude, a high index of suspicion is required to diagnose this entity early to reduce mortality.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Cyst infections in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2009;4:1183-9.
- [Google Scholar]




