

Translate this page into:
A Case of Joubert Syndrome with Chronic Kidney Disease
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Joubert syndrome is a genetically heterogeneous disorder that belongs to the group of cerebello-oculo-renal syndromes. It is characterised by neurodevelopmental abnormalities and complex midbrain-hindbrain malformation, visible on brain imaging as a molar tooth sign. It is classified as a ciliopathy and has variable renal involvement. Herein, we report a case of a 9-year-old boy with developmental delay, presented as chronic kidney disease and evaluation showed features of Joubert syndrome. Recognition of specific clinical and radiological findings will help in early diagnosis and appropriate care.
Keywords
Cerebello-oculo-renal syndrome
ciliopathy
Joubert syndrome
molar tooth sign

Introduction
Joubert syndrome (JS) is a rare autosomal recessive disorder, named after Marie Joubert in 1969.[1] It is a disorder of the midline structure of the brain having characteristic clinical and neuroradiological findings. Its prevalence is underestimated due to a lack of recognition of specific clinical signs and variable presentations. The diagnosis is based on the presence of the following three primary criteria: (1) the molar tooth sign on magnetic resonance imaging (MRI), (2) hypotonia in infancy and (3) developmental delay or intellectual disability. Renal involvement has been described in 30% of patients with Joubert syndrome.[2] In the present study, we report a case of JS presented with chronic kidney disease.
Case History
This is a 9-year-old boy [Figure 1], first offspring to 4th degree consanguineous marriage, born via preterm lower-segment caesarean section at 8 months of gestation in view of oligohydramnios and preterm premature rupture of membrane, with an uneventful antenatal period, and his birth weight was 2.2 kg. He had global developmental delay in attaining all milestones, with intellectual disability, generalised hypotonia of all muscles and congenital strabismus. Recently he was admitted to an outside hospital with fever and shortness of breath and in view of renal dysfunction, he was referred to our centre.

- Our patient with hypotonia and facial dysmorphism
On examination, he was found to have subnormal intelligence and he communicates with parents via gestures without proper word output. He has round facies, depressed nasal bridge, short philtrum, low set posteriorly rotated ears, brachydactyly, sandal gap due to second and third toe syndactyly. Biochemical profile showed anaemia (haemoglobin 6.4 g/dL), renal dysfunction (blood urea 157 mg/dL, serum creatinine 7.06 mg/dL), secondary hyperparathyroidism (serum calcium 6.0 mg/dL, serum phosphorus 6.1 mg/dL, serum PTH 322 pg/mL) and ultrasound abdomen showed normal-sized kidneys with increased echogenicity and loss of corticomedullary differentiation. In view of neurological abnormalities, MRI of the brain was taken, which showed vermian aplasia with characteristic molar tooth sign [Figure 2]. We treated the patient initially with six sessions of hemodialysis through right IJV dialysis catheter and later changed to continuous ambulatory peritoneal dialysis as patient had no suitable veins for AV fistula creation. Because of brain malformation he was started on prophylactic antiepileptic drug, levetiracetam. During hospital stay, he had status epilepticus refractory to conventional medical treatment, hence was intubated and started on mechanical ventilation as per neurologist opinion. Later developed ventilator-associated pneumonia and succumbed to illness. After taking consent from patient's father we performed a postmortem renal biopsy which showed 31 glomeruli, out of which 15 were sclerosed, viable glomeruli showed periglomerular fibrosis, interstitium was markedly fibrosed with scattered infiltrate of lymphocytes and plasma cells, and tubules were atrophic with focal thyroidisation and thickened basement membrane [Figure 3 and 4].

- Molar tooth sign in brain MRI
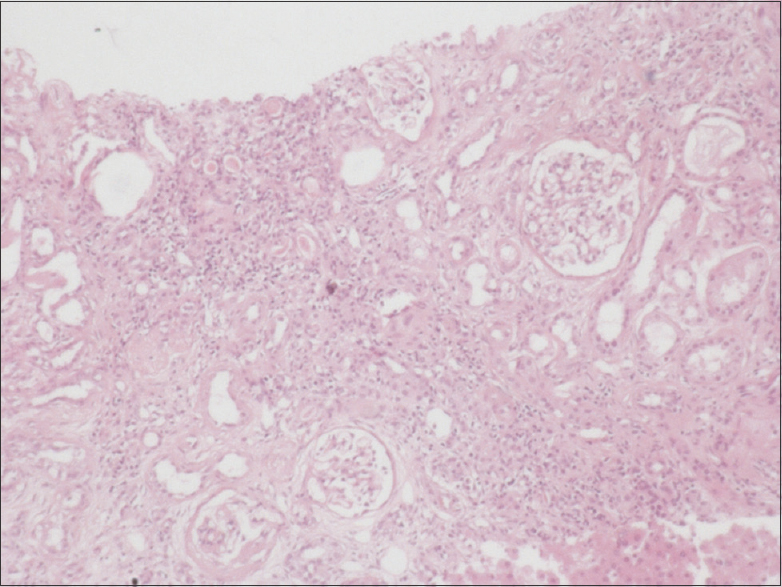
- Post-mortem renal biopsy shows tubular atrophy with focal thyroidisation and severe interstitial fibrosis
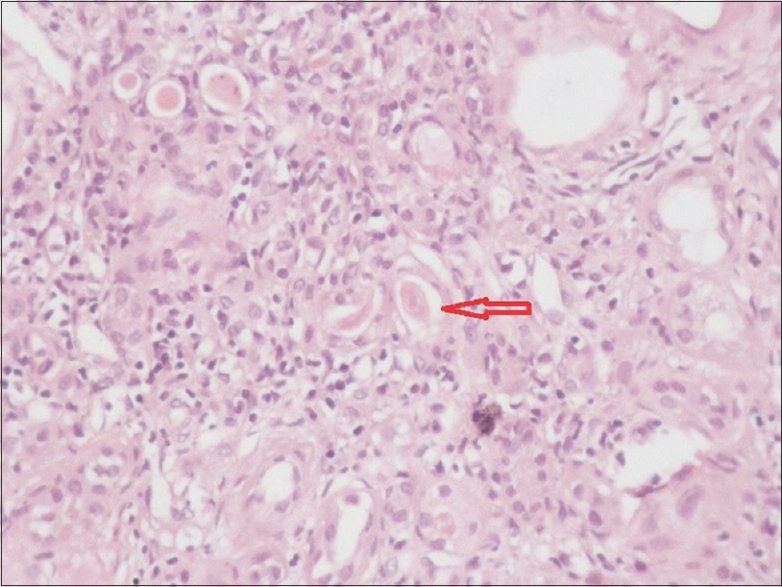
- Tubular atrophy with thyroidisation
Discussion
Joubert syndrome and related disorders (JSRD) are a genetically heterogeneous group of disorders with developmental delay and multiple other congenital anomalies, in which the characteristic MRI feature is the molar tooth sign. The average incidence of JSRD range between 1/80,000 and 1/100,000 live births.[3] These disorders have been classified as ciliopathies[4] and were attributed to >30 genes in which mutations in the AHI1, TMEM67 and CEP290 genes are common.[5] JSRD is classified into six different phenotypic subgroups: pure JS, JS with ocular defect, JS with renal defect, JS with oculo renal defects, JS with a hepatic defect and JS with orofaciodigital defects.[6].
The clinical features of Joubert syndrome include hypotonia, ataxia, abnormal eye and tongue movements, developmental delay and neonatal respiratory dysregulation (hyperpnoea). These may be associated with other organ involvement such as retinal dystrophy, hepatic fibrosis, nephronophthisis, polydactyly, cleft lip or cleft palate. The developmental delay and cognitive impairment cannot be fully explained by the hindbrain malformation and probably result from the involvement of the cerebral hemispheres. 'Molar tooth sign', due to its resemblance to the cross-section of a tooth on axial imaging, demonstrates deep posterior interpeduncular fossa, thick and elongated superior cerebellar peduncles, and hypoplastic or aplastic superior cerebellar vermis.[7].
Renal manifestations in Joubert syndrome are variable. It can occur as cystic dysplasia, polycystic kidney disease (PKD) or juvenile nephronophthisis. Nephronophthisis, which is common in Joubert syndrome, a form of chronic tubulointerstitial nephropathy, is characterised by normal or small-sized kidneys with increased echogenicity, lost corticomedullary differentiation and a few cysts at the corticomedullary junction. End-stage renal failure is usually reached by the end of the second decade, requiring renal replacement therapy. A characteristic feature of autosomal recessive PKD is early-onset severe hypertension and enlarged kidneys. PKD-nephronophthisis overlap pattern of kidney disease can also occur. Cystic dysplasia of the kidneys, which is often visualised on ultrasound imaging as multiple small cysts in small immature kidneys with foetal lobulations, and if associated with severe visual loss, is called Dekaban-Arima syndrome.[8] Urine osmolality[9] and neutrophil gelatinase-associated lipocalin (NGAL)[10] are considered as early indicators of renal damage in Joubert syndrome and help to start intensive nephrology follow-up.
Our patient with a history of developmental delay, intellectual disability and hypotonia, presented with chronic kidney disease requiring renal replacement therapy, on further evaluation found to have features of Joubert syndrome. Though there were no cysts in ultrasound imaging, kidneys were normal in size with increased echogenicity and loss of corticomedullary differentiation and postmortem biopsy showed tubular atrophy with the thickened basement membrane, scattered infiltrate and marked interstitial fibrosis (chronic interstitial nephritis). Hence, we assumed nephronophthisis as the probable cause of renal failure. Urine osmolality and NGAL (serum or urine) were not performed in our case as the patient was already presented in chronic kidney disease.
Conclusion
Joubert syndrome is a rare genetic disorder with characteristic neurodevelopmental and multiple other organ involvement. Nephronophthisis is a common renal manifestation. Detection of peculiar clinical and radiological findings helps in the appropriate diagnosis of various childhood syndromes.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813-25.
- [Google Scholar]
- Prospective evaluation of kidney disease in joubert syndrome. Clin J Am Soc Nephrol. 2017;12:1962-73.
- [Google Scholar]
- Mutation spectrum of Joubert syndrome and related disorders among Arabs. Hum Genome Var. 2014;1:14020.
- [Google Scholar]
- Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999;14:583-90.
- [Google Scholar]
- Cerebro-oculo-hepato-renal syndrome (Arima' syndrome): a distinct clinicopathological entity. J Child Neurol. 1986;1:338-46.
- [Google Scholar]
- Impaired urinary concentration ability is a sensitive predictor of renal disease progression in Joubert syndrome. Nephrol Dial Transplant 2018 doi: 10.1093/ndt/gfy333
- [Google Scholar]
- NGAL as an early biomarker of kidney disease in Joubert syndrome: Three brothers compared. Ren Fail. 2012;34:495-8.
- [Google Scholar]




