

Translate this page into:
IgA Nephropathy with Wilson's Disease: A Case Report and Literature Review
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The most common glomerulonephritis seen in the world is immunoglobulin A nephropathy (IgAN). It can be primary or secondary associated with various conditions like Chronic Liver disease, Crohn's disease, neoplasms, etc. However, IgAN secondary to Wilson's disease is very rare. A 9 year old boy presented with gross hematuria and proteinuria. He had a history of recurrent jaundice in the past. Ultrasonography (USG) whole abdomen showed altered echotexture of the liver with normal-sized kidneys. An extended workup for liver disease was done, and the diagnosis of Wilson's disease was confirmed with decreased serum ceruloplasmin levels, increased urinary copper, and the Kayser–Fleischer ring. Urine routine microscopy showed numerous red blood cells, few red blood cell casts, and mild proteinuria. Renal biopsy showed IgAN. The patient was started on D-penicillamine. On follow-up at 3 months, he showed complete resolution of proteinuria and hematuria. Thus, we suggest that Wilson's disease should be considered as one of the causes of secondary IgAN in pediatric patients with hematuria, proteinuria with liver dysfunction.
Keywords
Hematuria
IgA nephropathy
jaundice
KF ring
Wilson's disease

Introduction
IgA nephropathy is the most common form of primary glomerulonephritis in children and adults. Its incidence is at least 2.5 per 100,000.[1] It has a prevalence of 7.8', considering previous report from India.[2] Secondary IgAN can occur in association with liver diseases, infection-related, Inflammatory bowel disease, celiac disease, lung diseases, and neoplasm. The frequency of secondary IgAN in the pediatric age group is largely unknown. Liver disease is the most common cause of Secondary IgAN. The prevalence of hepatic IgAN has been reported in the range of 9–25' in various studies.[3456] Clinical, laboratory, or histopathology are generally indistinguishable from the primary IgAN. IgAN secondary to Wilson's disease is very rare. Herein, we report a case of a 9 year old boy with Wilson's disease presenting with gross hematuria and proteinuria with kidney biopsy showing IgAN.
Case Report
A 9-year-old male, born from non-consanguineous marriage, presented with two episodes of gross hematuria in the last 1 week. There was no complaint of fever, sore throat, arthralgia, myalgia, or abdominal pain. He had a history of two episodes of jaundice in the last two months. He had no family history of kidney or liver disease. His serum creatinine 2 months back was 0.93 mg/dL. His examination was significant for moderate pallor; there was no icterus. Systemic examination was unremarkable. Significant laboratory findings are shown in Table 1. Peripheral smear showed microcytic hypochromic anemia. The anemia profile was suggestive of iron deficiency anemia. His Coomb's test (direct and indirect) were negative. His estimated GFR was 62 mL/min/1.73 m2 (by using modified Schwartz formula). Differential diagnosis of secondary glomerulonephritis was sought. A workup for autoimmune disease was done, which was normal.
| Investigation | At admission | After 3 months of treatment |
|---|---|---|
| Hemoglobin (g/dL) | 9 | 12 |
| Total leukocyte count (cells per microliter) | 6000 | 5400 |
| Platelets (*103 (cells per microliter) | 130 | 220 |
| Total bilirubin (mg/dL) | 0.44 | 0.5 |
| SGOT (U/L) | 14 | 12 |
| SGPT (U/L) | 39 | 18 |
| ALP (U/L) | 76 | 54 |
| LDH (U/L) | 165 | 120 |
| Total protein (g/dL) | 6.64 | 6.2 |
| Albumin (g/dL) | 2.27 | 3.5 |
| PT/INR | 16.5/1.42 | 12/0.9 |
| Creatinine (mg/dL) | 0.93 | 0.82 |
| Serum HCO3- (mEq/L) | 25 | |
| C3 (mg/L) (970-1576) | 1384 | - |
| C4 (mg/L) (162-445) Viral serology: | 191 | - |
| Anti HAV | Negative | |
| Anti HEV | Negative | |
| HBsAg | Negative | |
| Anti HCV | Negative | |
| ANA | Negative | |
| ANCA | Negative | |
| ASMA | Negative | |
| Anti LKM | Negative | |
| 24 h urinary copper (<40) | 250 mcg/day | |
| Serum ceruloplasmin (20-40) | 5.4 mg/dL | |
| Urine routine microscopy | pH-6.7 | RBCs 1-2/HPF |
| 24 h | RBCs/HPF- Numerous RBC casts (+) Protein 2+Glucose negative | Protein (-) |
| Urine protein (g/d) | 1.1 | 0.21 |
| Calcium (100-300) | 138 mg/day | |
| Phosphorous (400-1300) | 565 mg/day |
Ultrasound abdomen was done, which showed normal-sized kidneys with mild liver atrophy, normal portal vein diameter (7.6 mm) without any splenomegaly or ascites. A kidney biopsy was performed, which showed 15 glomeruli. Mild mesangial proliferation was seen with no glomerulosclerosis, endocapillary proliferation, or crescent formation. Immunofluorescence study showed 3+ IgA along with a 1+ C3 deposit in the mesangium, thus, it was suggestive of IgA nephropathy with a MEST-C score of 1 [Figure 1].[78] In view of altered liver echotexture and recurrent jaundice in the past, a workup for liver disease was done including the viral as well as the autoimmune panel. Anti-hepatitis A virus, hepatitis E virus, hepatitis C virus, hepatitis B surface antigen were all negative. An autoimmune panel was also negative [Table 1]. There were no esophageal varices on upper GI endoscopy.
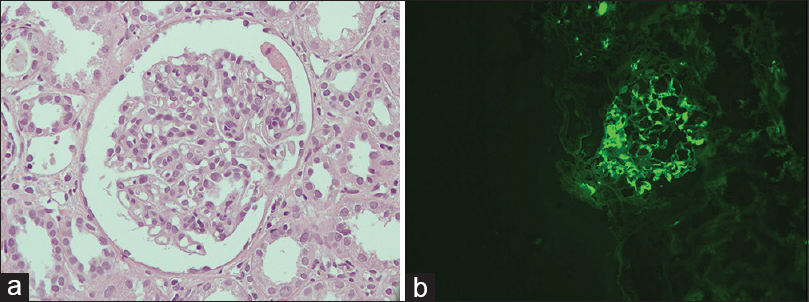
- (a) Glomerulus showing mesangial proliferation (H and E stain, original magnification ×400), (b) immunofluorescence showing mesangial granular deposits of IgA (original magnification ×200)
24 h urinary copper excretion was increased (250 mcg/day). The serum ceruloplasmin was low (5.4 mg/dL). The above findings were highly suggestive of Wilson's disease. Repeat physical examination revealed Kayser–Fleischer (KF) ring, visible on naked eye visualization, which was missed at first physical examination [Figure 2]. Thus, the combination of KF ring, low ceruloplasmin levels, and increased urinary copper levels established the diagnosis of Wilson's disease (Leipzig score 6; 4 or more points are require to establish the diagnosis). A detailed history was taken to rule out the neuropsychiatric features, and a thorough neurological examination was done for any tremors, ataxia, dystonia, etc., but there were no signs suggestive of neurological impairment. Urine biochemistry did not reveal any features of tubular damage [Table 1]. Liver biopsy and genetic testing could not be done.
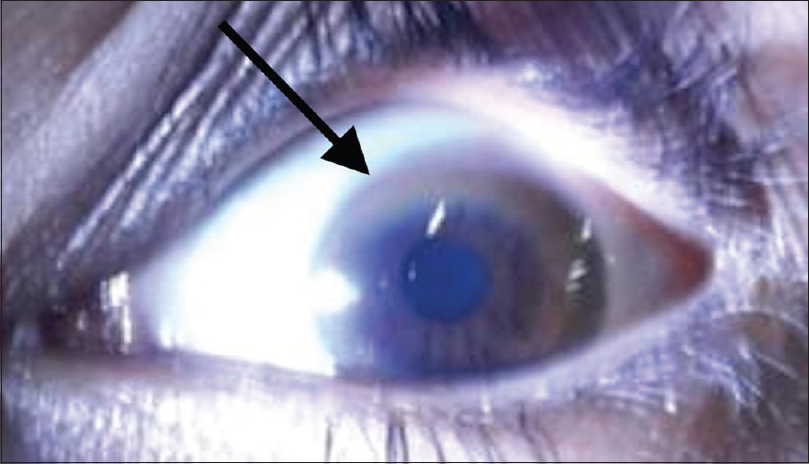
- Kayser-Fleischer ring (K-F ring) (black arrow) due to copper deposition on Descemet membrane visible on physical examination
The patient was started on d-penicillamine 200 mg twice a day with pyridoxine 20 mg once daily. He was observed for adverse reactions to d-penicillamine like diarrhea, skin rash, fever, and bone marrow suppression. Also, renal parameters and urine routine microscopy was done to look for disease activity and renal adverse effects secondary to the d-penicillamine. Repeat complete blood count (CBC), renal function test (RFT), transaminases, and urine analysis was done after 10 days of discharge, which showed persistent anemia with normal leukocyte count, normal RFT, and transaminases, urine analysis showed numerous RBC and 2+ albuminuria. He was advised to repeat these investigations on a monthly basis. Repeat 24-h urine copper was advised to check the adequacy of treatment but could not be done. He came for routine outpatient department follow-up after 3 months, which showed resolution of hematuria with a reduction in proteinuria (0.21 g/day) along with normalization of the anemia and liver function tests. Since penicillamine is a potentially nephrotoxic medication, we have planned to repeat the 24-h urine copper at 6 months and switch him on zinc acetate for maintenance therapy.
Discussion
Secondary IgAN is a broad term that includes the association of IgAN with various other systemic diseases. The pathogenesis, clinical course, response to treatment, and prognosis are highly variable among secondary IgAN. The factor which knit them together and distinguishes itself from primary IgAN is that immunosuppression is generally not advisable to treat the secondary IgAN. The pathogenesis of primary IgAN includes increased circulating galactose deficient IgA1 antibody. Antibodies directed against these galactose deficient IgA1 cause formation of immune complexes in the mesangium causing renal injury.[9] Several other hypotheses have been suggested for pathogenesis such as high levels of circulating IgA immune complexes, a reduction in the fractional catabolism of IgA and its complexes, and dysregulation of the IgA immune system; however, the precise pathogenesis remains uncertain.
The abnormal clearance of the circulating immune complexes by hepatic Kupffer cells may allow them to access the systemic circulation with subsequent deposition in the kidney.[10]
The renal outcome in hepatic IgAN is largely unknown. In a case series by Pouria S et al. reported the clinical renal outcome of biopsy proven hepatic IgAN over a period of 4 years. Four patients developed ESRD, two patients had progressive CKD, one had mild CKD, and one had normal renal functions.[11] However, the poor prognosis revealed in this report may be biased due to the fact that only those patients were biopsied who had a significant renal abnormality.
The resolution of secondary IgAN on the treatment of the underlying cause is variable. In a study by Hommos MS et al., they noticed that proteinuria and hematuria could persist even after liver transplant.[8] In another case, reported by Kalambokis et al., a 34-year-old man with cryptogenic liver cirrhosis with secondary IgAN had significant improvement in proteinuria after the treatment of portal hypertension.[11] Also, similar findings were noted with the case reported by Nakamura et al.[12] The variability among the outcome of hepatic IgAN is probably due to the degree of irreversible damage done to the kidneys before the primary disease is completely treated. In the present case, Wilson's disease was incidentally diagnosed while the patient was undergoing the evaluation for gross hematuria. His physical examination and laboratory investigation did not reveal any signs of liver decompensation or portal hypertension. He responded to the treatment in terms of improvement in liver and kidney parameters.
Wilson's disease is an autosomal recessive disease. It is characterized by copper accumulation in various tissue like the liver, eyes, and nerves. Gene for Wilson's disease (ATP7B) is also expressed in kidneys; thus, a variety of renal manifestations are seen. The frequency of renal manifestations is variable. Patients can have renal tubular dysfunction like proximal RTA, Fanconi syndrome causing loss of glucose, phosphate, amino acids, calcium, etc. Some patients present with hypercalciuria and nephrocalcinosis.[13] In a study by Zhuang et al., the authors specifically examined the association of renal abnormalities in pediatric patients with Wilson's disease.[14] Twenty-five patients (nine patients with renal dysfunction were excluded since they were on penicillamine) out of 85 individuals with Wilson's disease had some form of renal involvement. Five patients (5.8') had hematuria and proteinuria. One patient, whose renal biopsy report was available had features suggestive of IgAN. The outcome, in terms of renal survival, however, was not reported in this study.
IgAN associated with Wilson's disease is very rare, and only a few cases have been reported in the literature. A case reported by Sarles et al. found an association of cirrhosis of liver secondary to Wilson's disease with IgAN, however, treatment outcomes were not studied.[15] In the case reported by Tu J et al., a 9-year-old girl diagnosed to have IgAN after coming to medical attention because of gross hematuria.[16] After initiating treatment for Wilson's disease, within 2 months, her liver function improved along with the clinical remission of IgAN. Although we had used penicillamine in place of zinc sulfate, the result was similar in both cases. In the case report of an adult Wilson's disease with IgAN, reported by Shimamura et al., the patient was treated successfully with trientine hydrochloride and zinc acetate along with the improvement in his renal parameters.[17] The remission of IgAN with the treatment of Wilson's disease points towards its causation rather than just a chance association. However, since there was no kidney biopsy available after the remission, the determination of pathological remission of IgAN is still not entirely possible.
This report has a few limitations. First, liver biopsy and genetic testing could not be done. Secondly, A repeat 24 h urine copper was planned after the initiation of the therapy (an increase in excretion of copper above the baseline is suggestive of the adequacy of treatment). However, the patient took discharge after 2 days of the initiation of the treatment due to personal reasons.
In conclusion, IgAN secondary to Wilson's disease is rare, but an important and treatable cause of secondary IgAN in pediatric patients. Wilson's disease should be considered as the differential diagnosis of secondary IgAN in pediatric patients with hematuria, proteinuria with liver dysfunction.
Informed consent
Written informed consent was obtained from the patient to report this case.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- IgA nephropathy in a tertiary care center from south India. Indian J Nephrol. 2011;21:230-4.
- [Google Scholar]
- Renal glomerular lesions in unselected patients with cirrhosis undergoing orthotopic liver transplantation. Pathology. 1995;27:237-46.
- [Google Scholar]
- Brief communication: Glomerulonephritis in patients with hepatitis C cirrhosis undergoing liver transplantation. Ann Intern Med. 2006;144:735-41.
- [Google Scholar]
- Renal outcomes in patients with IgA nephropathy undergoing liver transplant: A retrospective cohort study. Transplant Direct. 2017;3:e193.
- [Google Scholar]
- Prospective assessment of renal histopathological lesions in patients with end-stage liver disease: Effects on long-term renal function after liver transplantation. J Hepatol. 2012;57:572-6.
- [Google Scholar]
- Oxford classification of IgA nephropathy 2016: An update from the IgA nephropathy classification working group. Kidney Int. 2017;91:1014-21.
- [Google Scholar]
- Association of liver cirrhosis related IgA nephropathy with portal hypertension. World J Gastroenterol. 2007;13(43):5783-6.
- [Google Scholar]
- IgA nephropathy associated with portal hypertension in liver cirrhosis due to non-alcoholic and non-A, non-B, non-C hepatitis. Intern Med. 1994;33:488-91.
- [Google Scholar]
- Analysis of renal impairment in children with Wilson's disease. World J Pediatr. 2008;4:102-5.
- [Google Scholar]
- Wilson disease, IgA glomerulonephritis and vascular purpura: An incidental association? Arch Fr Pediatr. 1993;50:501-4.
- [Google Scholar]
- A special case of recurrent gross hematuria: Answers. Pediatr Nephrol. 2017;32:273-5.
- [Google Scholar]
- Immunoglobulin A nephropathy secondary to Wilson's disease: A case report and literature review. CEN Case Rep. 2019;8:61-6.
- [Google Scholar]




