

Translate this page into:
An Unusual Clinical Manifestation of Plasmablastic Lymphoma in a Renal Transplant Recipient
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Plasmablastic lymphoma is a rapidly progressive CD20 negative large cell non-Hodgkin lymphoma with poor outcome. It occurs mostly in immunocompromised individuals and has a predilection for extranodal sites. They need to be differentiated from other entities sharing similar morphological features like poorly differentiated carcinoma, Burkitt's lymphoma, Alk positive large B cell lymphoma, Diffuse large B cell lymphoma, and anaplastic myeloma. EBV negativity in recipients, type, intensity, and duration of immunosuppressives used are certain risk factors in development of posttransplant lymphoproliferative disorders. High index of suspicion can help clinch the diagnosis early and prevent catastrophic consequences. Our renal transplant recipient presented with complaints of pain abdomen and malena for which he underwent exploratory laparotomy. Diagnosis was established on histopathology and timely treatment initiated reverted the disease.
Keywords
Immunosuppression
plasmablastic lymphoma
post transplant lymphoproliferative disorder
solid organ transplantation

Introduction
Plasmablastic lymphoma (PBL) is a rare, aggressive B-cell non-Hodgkin lymphoma with immunoblastic and plasmacytoid morphology and poor response to chemotherapy. It was first described way back in 1997 by Delecluse et al.[1] in patients with HIV. They are now known to be associated with use of increased immunosuppression, post solid organ transplant, patients with underlying autoimmune disease, chronic inflammatory state and in elderly.[2] PBL has primarily extranodal involvement with common sites being oral cavity, gastrointestinal tract, skin, retroperitoneum, and soft tissue. Patients with advanced clinical stage may also involve the bone marrow. Lymph node involvement is less frequent at diagnosis however, it has been described in up to 30% of posttransplant patients.[3] Most cases of PBL show Epstein Barr virus (EBV) positivity suggesting its role in disease pathogenesis. We hereby report a renal transplant recipient who presented 08 years after transplant with complaints of abdominal pain and malena.
Case Details
A 35-year-old male patient with end-stage renal disease (ESRD) due to IgA Nephropathy underwent live-related ABO compatible renal transplant, with father as donor in 2012, 3 months after being initiated on maintenance hemodialysis. Donor was 53-year-old with donor kidney Glomerular filtration rate (GFR) of 51.6 mL/min and was haploidentical match with recipient. Pre-transplant immunological workup included panel reactive antibody (PRA) against class I and class II HLA antigens which was 0%, complement dependent cytotoxicity crossmatch (CDC) and flow-cytometry crossmatch which were negative. Infectious monitoring for EBV and CMV status was as D+/R+. Patient received induction therapy with Injection Basiliximab 20 mg on D + 0 and D + 4 and maintenance was with tacrolimus, mycophenolate mofetil, and prednisolone. He had stable course and suffered no rejection episodes or opportunistic infections. He presented eight years after renal transplant with history of pain abdomen and malena. At presentation, he had a haemoglobin of 6.0 g/dL (normal range 13.0-18.0 g/dL), MCV- 80 fL and his stool for occult blood was positive. Upper GI endoscopy done was normal, however colonoscopy showed multiple large oval ulcers in cecum with white exudates at base and friable edges with normal intervening mucosa. Multiple smaller ulcers of variable sizes were also seen in ascending colon and proximal transverse colon with clean base. Histopathologic examination of ulcer showed no crypt destruction, cryptitis or crypt abscess, granulomas or atypia. Tissue polymerase chain reaction (PCR) for Cytomegalovirus (CMV) was positive whereas his serum EBV PCR was positive. He was initially managed as CMV colitis with Injection Ganciclovir followed by Valganciclovir for a total of 03 weeks with supportive measures. He continued to be symptomatic and required a total of 11 packed RBC transfusions. A contrast-enhanced computed tomography scan of abdomen showed dilation of stomach, duodenum, and proximal jejunal loops with intussusception lesion causing partial obstruction. Patient was taken up for exploratory laparotomy with resection of partial segment of jejunum. On gross examination, there was an intussuscepted loop identified with a large growth measuring 8.5 cm × 6.5 cm × 3.0 cm [Figure 1]. Histopathologic examination from mass showed normal jejunal mucosa with focal denudation. The underlying submucosa showed diffuse infiltration by sheets of monomorphic large atypical lymphoid cells with immunoblastic morphology [Figure 2a and b]. On immunohistochemistry the tumor cells were strongly positive for CD38, CD138 with Ki67 labeling of 90%. The tumor cells were negative for CD20, CD3, CD56, CD34 [Figure 3a and b]. A positron emission tomography (PET) scan done after surgery showed fluorodeoxyglucose (FDG) avidity only at operative site likely due to post-op inflammatory change. CSF was negative for blast cells. Bone marrow aspiration and biopsy was normal. He was started on chemotherapy with Cyclophosphamide, doxorubicin, vincristine, high dose prednisolone (CHOP regime), lenalidomide, and CNS prophylaxis with intrathecal methotrexate. His immunosuppression was reduced. Mycophenolate mofetil was tapered and stopped. mTOR agents in the form of everolimus were added as the third immunosuppressant and the dose of calcineurin inhibitors was reduced after explaining the risk of graft rejection. A repeat PET scan after four cycles of chemotherapy showed no FDG avid lesions anywhere in body. He maintains good functional status and normal renal function.
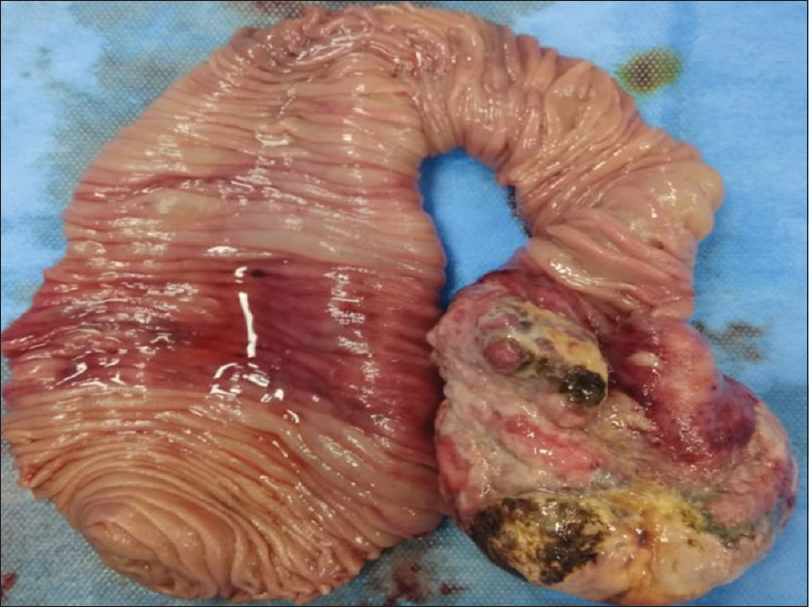
- A large growth measuring 8.5 × 06 × 2 cm is seen at one end of resected jejunal loop which is projecting into lumen. The lining mucosa is focally ulcerated
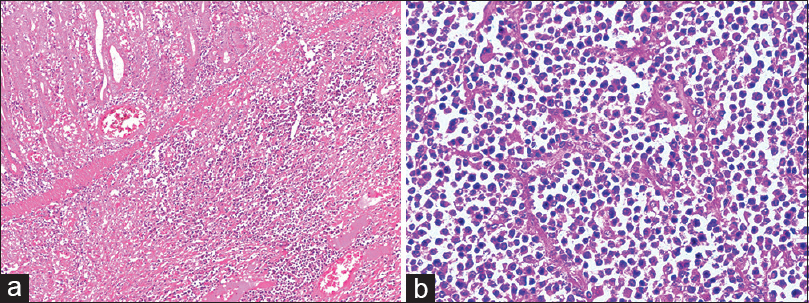
- (a) The Hematoxylin and Eosin (H and E; 100×) sections from the tumor mass showed a normal mucosal lining with infiltration of underlying submucosa by sheets of large monomorphic atypical lymphoid cells having immunoblastic morphology. (b) At 400 × magnification individual tumor cells have large vesicular nucleus with prominent nucleoli and eosinophilic cytoplasm. Interspersed are seen numerous plasma cells with frequent binucleate forms

- (a and b) Immunohistochemical expression showing strong cytoplasmic positivity for CD138 and nuclear Ki67 proliferative index of 90% in the atypical lymphoid cells
Discussion
PBL is a monomorphic posttransplant lymphoproliferative disorder (PTLD) that displays immunoblastic/plasmablastic morphology, has an aggressive clinical course and shows poor survival. Since its first description in HIV positive patients, they are now known entity in nonHIV background. PBL presenting as a cutaneous leg ulcer, post solid organ transplant was first reported in 2003. Since the initial report large number of cases have been published over the years in transplant recipients. There are numerous risk factors causing PTLD. Among them the most important component implicated are EBV seronegativity in recipient, type of organ transplant, intensity and duration of immunosuppressive agents administered.[45]
Our patient received induction with Basiliximab (IL-2 receptor antagonist) placing him at risk for lymphomagenesis. The role of EBV in development of PTLD's (including plasmablastic lymphoma) is well established as they cause transformation and immortalization of B cells leading to uncontrolled proliferation. Besides, molecular studies in PBL have revealed complex cytogenetic abnormalities. Most common alteration seen is either translocation of MYC oncogene with IG or overexpression of MYC.[6] Rarely mutation in PRDM1, a gene that encodes for BLIMP1 protein, a transcriptional repressor has been reported in these lymphomas. Any or both these molecular aberrations leads to transformation of activated B cells in the germinal center and their uncontrolled growth.
The most common site of involvement by PBL in posttransplant nonHIV cases as reported is lymph node and skin followed by GIT tract as against HIV patients having high predilection for oral cavity.[7] Komaranchath et al.[8] in their case series on PBL involving GI tract have shown distal GI region predilection. Our case was interesting as patient presented with features of intestinal obstruction due to a large jejunal mass. Histologically PBL is characterized by diffuse proliferation of large lymphoid cells having central round nuclei with prominent nucleoli and abundant amphophilic cytoplasm. The nuclei may be eccentrically located giving a plasmablastic appearance. Immunophenotypically the neoplastic cells are negative for B cell markers including CD19, CD20, and PAX5 and positive for markers of plasma cell neoplasm like CD79a, CD38, CD138, and MUM1. MYC and PRDM1 (Blimp1) may be detected in subset of patients.[9]
PBL simulates anaplastic myeloma both morphologically and immunophenotypically. Anaplastic myeloma; however, has distinct clinical, radiological, and laboratory findings. Features favoring PBL include strong EBV encoded RNA (EBNA) positivity in a known immunocompromised individual. ALK-positive large B-cell lymphoma bears much similarity to PBL; however, it shows ALK positivity and characteristic translocation. Another differential to be considered is Primary effusion lymphoma that presents with serous effusions and has universal association with human herpesvirus 8. The treatment options for PBL, posttransplant remains varied and heterogeneous and includes reduced immunosuppression, chemotherapeutic regimens like CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), more intensive protocols with addition of etoposide and prophylactic intrathecal methotrexate. Immunomodulatory anti-myeloma drug lenalidomide and use of anti protease inhibitor Bortezomib have shown promising results. Our patient received it in combination with CHOP therapy and the repeat PET scan done after four cycles showed complete disease resolution. Presently, our patient is disease free and is on regular follow-up at nephrology and medical oncology outpatient department.[101112]
Conclusion
PBL is a rare large B-cell lymphoma with dismal outcomes and poor response to therapy. From the initial description in patients of HIV, PBL is now reported in all immunocompromised patients especially post solid organ transplant. Awareness of this aggressive entity leads to early diagnosis and therapeutic intervention with prolonged remissions. Role of immunotherapy, immunomodulatory agents, and drugs against specific molecular or signaling pathways involved in lymphomagenesis are being explored.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his clinical information to be reported in the journal. The patient understands that his name and initials will not be published, and due efforts will be made to conceal his identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Plasmablastic lymphomas of the oral cavity: A new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413-20.
- [Google Scholar]
- Clinicopathologic comparison of plasmablastic lymphoma in HIV-positive, immunocompetent, and posttransplant patients: Single-center series of 25 cases and meta-analysis of 277 reported cases. Am J Surg Pathol. 2014;38:875-86.
- [Google Scholar]
- Stage, age, and EBV status impact outcomes of plasmablastic lymphoma patients: A clinicopathologic analysis of 61 patients. J Hematol Oncol. 2015;8:65.
- [Google Scholar]
- Impact of Epstein–Barr virus donor and recipient serostatus on the incidence of post-transplant lymphoproliferative disorder in kidney transplant recipients. Nephrol Dial Transplant. 2012;27:2971-9.
- [Google Scholar]
- The impact of EBV status on characteristics and outcomes of posttransplantation lymphoproliferative disorder. Am J Transplant. 2015;15:2665-73.
- [Google Scholar]
- Advances in the understanding of MYC-induced lymphomagenesis. Br J Haematol. 2010;149:484-97.
- [Google Scholar]
- Plasmablastic lymphoma of the gastrointestinal tract: A rare entity with dismal prognosis. Indian J Cancer. 2016;53:529-33.
- [Google Scholar]
- Plasmablastic lymphomas and plasmablastic plasma cell myelomas have nearly identical immunophenotypic profiles. Mod Pathol. 2005;18:806-15.
- [Google Scholar]
- Bortezomib in combination with infusional dose-adjusted EPOCH for the treatment of plasmablastic lymphoma. Br J Haematol. 2015;169:352-5.
- [Google Scholar]
- A case of plasmablastic lymphoma achieving complete response and durable remission after lenalidomide-based therapy. Oncol Rest Treat. 2017;40:46-8.
- [Google Scholar]
- Plasmablastic posttransplant lymphoma: Cytogenetic aberrations and lack of Epstein-Barr virus association linked with poor outcome in the prospective German Posttransplant Lymphoproliferative disorder registry. Transplantation. 2012;93:543-50.
- [Google Scholar]




