

Translate this page into:
An Unusual Presentation of Multiple Myeloma
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Multiple myeloma commonly presents as anemia, renal failure, bone pain, and infections. Presentation with epistaxis is extremely rare, and hence myeloma as the etiologic factor is seldom considered. We report the case of a patient who initially presented with recurrent epistaxis and then with myasthenia. It was only when he developed acute kidney injury 4 months after the initial presentation with epistaxis that a diagnosis of multiple myeloma was made.
Keywords
Acute kidney injury
epistaxis
multiple myeloma
myasthenia

Introduction
Multiple myeloma (MM) is a malignant proliferation of plasma cells producing a monoclonal paraprotein in the bone marrow. It accounts for 10% of all hematologic malignancies, and renal failure occurs in 20% to 40% of newly diagnosed patients with MM. It can present in a variety of ways. In 70% of patients, the most common symptoms are back or bone pain, fatigue, and recurrent or persistent infections. It can also present with symptoms of hyperviscosity, neuropathy, shortness of breath, and emergency medical problems such as vertebral compression fractures and damage to the nerves of the spinal cord. Presentation with epistaxis and myasthenia is extremely rare. We report one such rare case.
Case Description
A 52-year-old male, known case of ischemic heart disease (angioplasty in 2002) for which he was on Aspirin tablet 75 mg, developed bleeding from the nose in February 2019. He went to an ENT (ear, nose, and throat) surgeon who advised nasal packing and conservative management. His blood pressure was 130/90 mmHg at that time. In March 2019, he developed breathlessness on exertion for which he went to a cardiologist who ordered an electrocardiogram (ECG), two-dimensional echocardiography, and X-ray chest that were normal. Complete blood count revealed hemoglobin 12.6 g/dL, white blood cell 5,590 cells/mm3, and platelet count 262,000 cells/μL. He then went to a pulmonologist who advised contrast-enhanced computed tomography (CT) of the chest, which showed findings of small airway disease. An incidental finding reported was a collapse of the C7 vertebrae. This, it appears, was completely missed. He was prescribed salbutamol metered dose inhaler and theophylline. His complaints were still persistent, so he consulted a general physician in April 2019, who advised certain tests. Urinalysis revealed 2+ protein, trace blood, 2 to 4 red blood cells/high-power field, and 6 to 8 white blood cells/high-power field. Complete blood count revealed hemoglobin 12 g/dL, white blood cell 7,000/mm3, platelet count 269,000/μL, and serum creatinine 0.82 mg/dL. Ultrasound abdomen was normal. No further workup was done for urinary abnormalities.
In May 2019, there was a progressive change in voice on exertion, which used to get better with rest. There was no involvement of any other muscles. He consulted a neurologist who diagnosed his acute bulbar weakness clinically as myasthenia gravis. His acetylcholine receptors (AchR) antibody was negative, and the repeated nerve stimulation test (RNST) was negative. He was given three doses of 500 mg of methylprednisolone followed by a tapering dose of deflazacort and started on tablet pyridostigmine 60 mg thrice a day. He continued the same medications with partial relief of symptoms.
In June 2019, he complained of burning micturition. There was no fever, hematuria, or flank pain. Urinalysis revealed 1+ protein and >200 white blood cells/high-power field. He was treated with Amikacin injection 500 mg, 12 hourly for 7 days. During that period, he had two episodes of melena, so oesophago-gastro-duodenoscopy was done, which showed antral gastritis. After 1 week of antibiotic, a repeat urinalysis revealed 4+ protein, 0 to 2 red blood cells/high-power field, and 2 to 3 white blood cells/high-power field. Urine albumin to creatinine ratio was 408 μg/g. His creatinine was 12.6 mg/dL.
With the above history and a significant weight loss of 20 to 25 kg over 5 months, the patient was referred to our unit. On admission, he was hemodynamically stable, and general and systemic examination was unremarkable. Routine blood and urine tests revealed the following results: Urinalysis revealed 2+ protein, 8 to 10 white blood cells/high-power field, and 4 to 5 red blood cells/high-power field. Spot urine protein creatinine ratio was 5.67. Complete blood count revealed hemoglobin 8.3 g/dL, white blood cell 8,470 cells/mm3, and platelet count 281,000 cells/μL. Serum creatinine was 15.91 mg/dL, blood urea nitrogen 59 mg/dL, sodium 132 mEq/L, potassium 5.7 mEq/L, bicarbonate 17 mEq/L, calcium 9.2 mg/dL, phosphorous 8.56 mg/dL, total protein 6 g/dL, albumin 3.4 g/dL, globulin 2.6 g/dL, alkaline phosphatase 119 U/L, uric acid 1.18 mg/dL, and total CPK (creatine phosphokinase) 16 U/L. Differential diagnosis considered at this time was acute tubular necrosis due to aminoglycoside toxicity, rapidly proliferative glomerulonephritis, or paraproteinemia. Serum C3 complement level was normal. Serum antinuclear antibodies, antimyeloperoxidase, antiproteinase 3, and anti–glomerular basement membrane antibodies were negative. Serum protein electrophoresis was sent, and a kidney biopsy was performed after two sessions of hemodialysis in view of marked azotemia.
Serum protein electrophoresis (SPE) showed an M-spike with M-protein of 0.17 g/dL in the kappa region. Serum agar gel immunofixation electrophoresis showed the presence of monoclonal immunoglobulin IgG class with kappa light chain. Serum free kappa light chain was 5,100 mg/L (3.3–19.4), and free lambda light chain was 12.1 mg/L (5.71–26.3) with a ratio of 421.49 (0.37–3.1).
Renal biopsy showed 20 glomeruli, and 6/20 glomeruli showed a mild increase in mesangial cellularity. No crescents or organized deposits were seen. Many of the terminal tubules showed granular eosinophilic focally lamellate, PAS (periodic acid–Schiff) negative, and fractured casts with sloughing of tubular epithelium. Intratubular calcification was also noted. Mild tubular atrophy was seen. Interstitium showed edema, focal collections of lymphocytes, and few neutrophils with mild areas of fibrosis. Vessels showed mild thickening. There was no evidence of vasculitis. Immunofluorescence microscopy showed that tubular casts stained 3 + for kappa light chains and were negative for lambda light chains [Figure 1].
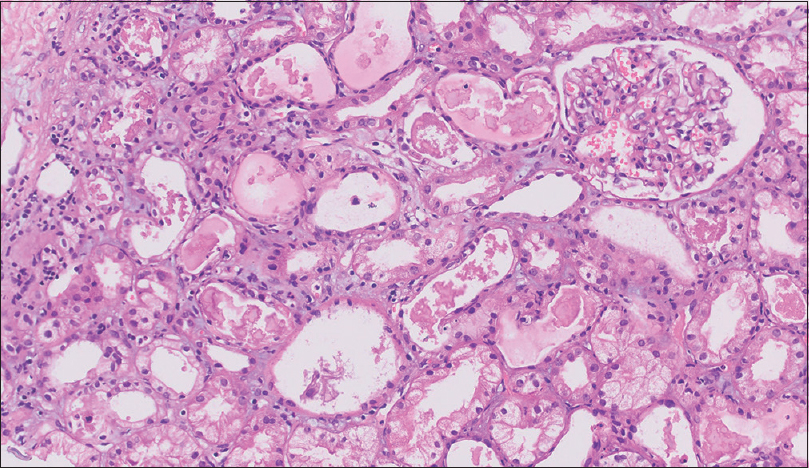
- Renal biopsy showing the terminal tubules having granular eosinophilic focally lamellate, PAS negative, and fractured cast with sloughing of tubular epithelium. Interstitium showed edema, focal collections of lymphocytes, and few neutrophils with mild areas of fibrosis
On further evaluation, there was no evidence of lytic lesions on the skeletal survey by X-ray. Bone marrow trephine biopsy showed moderately cellular marrow, minimal fibrosis, and increased plasma cell population 60% to 65% with a kappa monoclonal expression on immunohistochemistry. Fluorescence in situ hybridization analysis showed negative status for immunoglobulin H gene rearrangement. Both myeloid and erythroid cell lines were reduced. The constellation of all these features and investigations were consistent with a diagnosis of kappa light chain MM with light chain cast nephropathy.
The patient was started on steroids and bortezomib. Hemodialysis was continued for his acute kidney injury. In July 2019, he came off dialysis. His serum kappa free light chain had decreased to 562 mg/L. His myasthenic symptoms completely improved after starting treatment of MM. His creatinine was 1.2 mg/dL at his last follow-up.
Discussion
MM accounts for 1% of all neoplastic disorders and 10% of all hematologic malignancies.[1] The incidence increases with increasing age.
Most patients with MM present with anemia, renal failure, bone pain, or pathologic fractures of the axial skeleton. Even in this case, when the patient presented with breathlessness on exertion to his physician and contrast-enhanced CT chest was done, collapse of the C7 vertebrae was reported. However, this was missed by the treating physician.
In approximately 30% of cases, MM is discovered through routine blood testing when patients are being evaluated for unrelated problems. Neurologic symptoms and hyperviscosity are uncommon presentations.[2]
This patient presented initially to his doctor with epistaxis. In most cases (70%–80%), the cause of epistaxis is unknown, and bleeding is self-limiting. Epistaxis as a presentation of MM is extremely rare.[3] Although qualitative platelet dysfunction due to paraprotein is probably the most common mechanism, the pathophysiology may be multifactorial, including thrombocytopenia, uremia, hyperviscosity, inhibition of fibrin monomers polymerization, circulating heparin-like anticoagulant, Factor X deficiency, acquired von Willebrand syndrome, local tissue fragility secondary to amyloidosis, and vascular endothelium damage. This patient had no thrombocytopenia and no uremia when he presented with epistaxis. The cause of the bleeding may be qualitative platelet dysfunction or interference with coagulation factors.[2]
Another uncommon feature with which this patient manifested is myasthenia gravis, which is an autoimmune disease resulting from antibodies that block or destroy nicotinic AchR at the junction between the nerve and the muscle. Myasthenia gravis can present as isolated bulbar weakness in which AchR antibody can be negative. RNST can be negative because laryngeal muscles were not checked during the test.[4] The association of myasthenia gravis and MM was first reported by Rowland et al.[5] who diagnosed a plasma cell dyscrasia in a patient 5 months after the diagnosis of MG.
Several cases of autoimmune conditions related to MM are documented in the literature. In the majority of cases, the diagnoses are made simultaneously; systemic lupus erythematosus is the most common autoimmune disorder associated with MM. The mechanism of association between autoimmune disorders and myeloma is not known. According to a few studies, autoimmunity favors the escape of abnormal clones of B cells from regulatory mechanisms leading to the emergence of neoplastic B cell clones.[6]
In summary, this patient presented with two very unusual but known features: epistaxis and myasthenia gravis. Knowing that in 30% of cases MM is discovered through routine blood testing when patients are being evaluated for unrelated problems, SPE and serum free light chains should be a part of the evaluation in those who fall in the age-group of the patient presented.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Multiple myeloma: Clinical features and indications for therapy. Best Pract Res Clin Haematol. 2005;18:553-68.
- [Google Scholar]
- The otolaryngologic manifestations of multiple myeloma. Laryngoscope. 1978;88:1320-32.
- [Google Scholar]
- Respiratory assessment of myasthenia gravis patients using repetitive nerve stimulation of phrenic and intercostal nerves. Neurol India. 2020;68:1394-9.
- [Google Scholar]
- Myasthenia gravis with a myeloma-type, gamma-G (IgG) immunoglobulin abnormality. Am J Med. 1969;46:599-605.
- [Google Scholar]
- Autoimmune disorders and multiple myeloma-two illustrative case reports and a literature review. Rev Colomb Cancerol. 2018;22:76-83.
- [Google Scholar]




