

Translate this page into:
Bardet–Biedl Syndrome Presenting in Adulthood
Address for correspondence: Dr. Bhavik Prajapati, C203, Aastha Emerald, Near Neelkanth Mahadev Temple, Behind Rannapark Bus Stand, Naranpura, Ahmedabad – 380 013, Gujarat, India. E-mail: bhavikap87@yahoo.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Bardet–Biedl syndrome (BBS) is a rare disorder with a frequency of 1:1,60,000. The disease is inherited in an autosomal recessive manner. Less than 15 cases have been reported from India. We present a case of Bardet–Biedl syndrome presenting to the medical emergency with acute breathlessness because of de-compensated renal failure and salient features such as marked polydactyly, central obesity, retinitis pigmentosa, end-stage renal diseases, and mental retardation. Genetic study showed that the patient had BBS genetic variant 9 (MIM#615896), VUS variant. The patient was primarily treated for end-stage chronic renal failure with hemodialysis. We are reporting this case for its rarity and the presence of a novel genetic variant of an unidentified significance as per genome mapping. BBS is often not diagnosed at all or diagnosed late until end-stage renal failure sets in. Timely diagnosis might not help treat the condition but surely improve the quality of life for the patient.
Keywords
Bardet–Biedl syndrome
central obesity
chronic renal failure
polydactyly
retinal dystrophy

Introduction
Bardet–Biedl syndrome (BBS) is an uncommon autosomal recessive disorder characterized by post-axial polydactyly, central obesity, retinal dystrophy, hypogonadism, learning disabilities, and renal abnormalities.[1] The Bardet–Beidl Syndrome phenotype has been associated with 16 different genes.[1] The syndrome has a frequency of 1:1,60,000.[2] Less than 15 cases have been reported from India.[3] We present a classic case of Bardet–Biedl syndrome presenting to the medical emergency with acute breathlessness because of renal failure and salient features such as marked polydactyly, central obesity, retinitis pigmentosa, end-stage renal diseases, and mental retardation. A genetic study was performed, which showed that the patient had BBS genetic variant 9 (MIM#615896) – VUS variant.
Case History
A 32-year-old, unmarried, female, belonging to lower socio-economic class, born of consanguineous marriage, was brought to the medical emergency with painless, pitting bipedal edema extending up to the level of the knee, facial edema since the past 1 month [Figure 1a], and acute onset of breathlessness at rest, not associated with the positional or diurnal variation, since 1 day. She had a regular menstrual cycle. Her menarche set in at the age of 14 years. The patient also had complete loss of vision in both eyes since 9 years of age and bronchial asthma since 7 years of age.

- (a) Facial oedema due to renal compensation. (b) Malocclusion of teeth (c) Central Obesity
She had a temperature of 98°F, a pulse rate of 100/min, a blood pressure of 160/100 mm of Hg, a respiratory rate of 24/min, the random blood sugar of 143 mg/dl, and an oxygen saturation of 94% on room air. A detailed general examination of the patient was performed. The patient had a body mass index (BMI) of 30.2 kg/m2 (Obese Class 1). She had evident central obesity. Her left eye showed exotropia. Malocclusion of teeth was also noted [Figure 1b]. There was post-axial polydactyly with hexadactyly in the left upper limb and both lower limbs [Figure 2c].
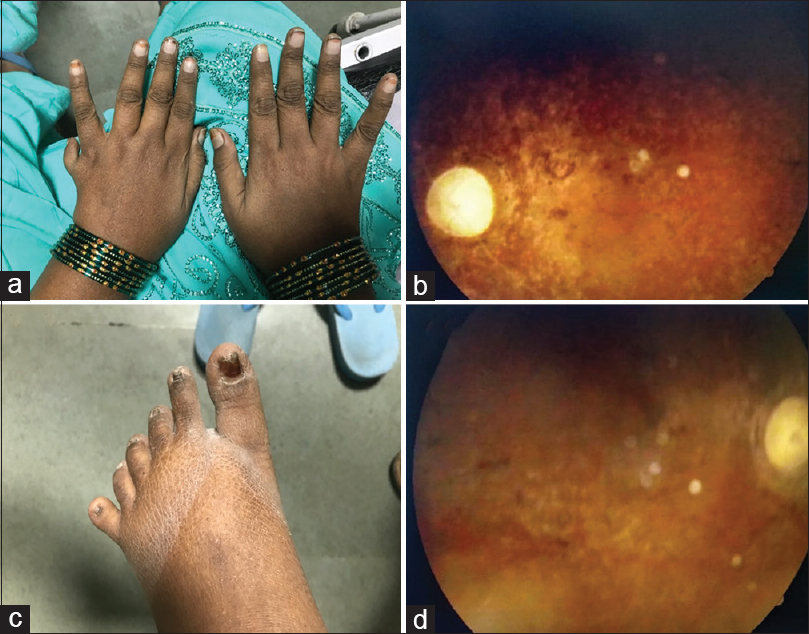
- (a) Hexadactyly in the left hand (b) Retinitis Pigmentosa on fundoscopy (c) Hexadactyly in lower limb (d) Retinitis Pigmentosa on fundoscopy
Cardiovascular and neurological examination was normal. On respiratory auscultation, coarse crepitations were heard in bilateral lower lung zones. Mini–Mental State Examination blind[4] totalled up to 11/22. A visual acuity test was suggestive of complete blindness with no perception of light in both eyes. Fundoscopy revealed a pale waxy disc with bony spicules, suggestive of retinitis pigmentosa [Figure 2d].
She had a urine output of 800 ml/24 hours. Chest X-ray was suggestive of pulmonary edema and moderate left pleural effusion without any pneumonitis.
The hemogram showed no abnormality, but the renal function test was markedly altered – S. Urea – 208 mg/dl, S. Creatinine – 7.20, S. Calcium was low (5.2 mg/dl), inorganic phosphate was high (8.2 mg/dl), and S. Uric acid was also high (10.3 mg/dl). The serum intact parathyroid level was elevated (753 pg/ml). Urine examination showed positivity for protein (30 mg/dl), blood (0.75 mg/dl), albumin (150 mg/L), isomorphic red blood cells, white blood cells [101 cells/high power field (HPF)], and bacteria (20,000 bacteria/HPF). The protein creatinine ratio was >=0.50 g/g. The albumin creatinine ratio was >=300 mg/g. Creatinine clearance by the Crockcroft–Gold formula was calculated to be 12 ml/min. The estimated glomerular filtration rate by the Modification of Diet in Renal Disease equation came out to be 7 ml/min/1.73 m2, indicating Stage V chronic kidney disease according to KDIGO Guidelines 2012.[5] Ultra-sonography of the abdomen showed a bilaterally small-sized kidney with loss of cortico-medullary differentiation (left kidney size – 6.1 cm × 2.4 cm and right kidney size – 6.5 cm × 3.1 cm).
A genetic panel study for Bardet–Biedl syndrome was performed. Homozygous missense variant c.711 G>T (p.W237C) of BBS9 at Exon 8 of Chromosome 7 (Chr7:33312632) was mapped in our patient's genome. The variant is a VUS-type variant.
The patient was primarily treated for end-stage chronic renal failure. An immediate 3.5 hours of hemodialysis with an ultra-filtration rate of 6.3 ml/Kg/hour was performed after the patient's altered renal function test was obtained. Appropriate antibiotic coverage was given. Pleural effusion and breathlessness resolved after dialysis indicating reduced volume overload. The patient was put on maintenance hemodialysis twice/week and discharged. No family screening was performed because of financial constraints of the patient. Her family members were counselled and called for a regular follow-up.
Discussion
In lieu of the patient being a product of consanguineous marriage and the presence of central obesity, strabismus, malocclusion of teeth, complete blindness because of retinitis pigmentosa, post-axial polydactyly with hexadactyly, mental retardation, and end-stage chronic renal failure, she was suspected to have Bardet–Beidel syndrome. Our patient had all the features except hypogonadism.
Bardet–Beidel syndrome is diagnosed in childhood, but our case is of a 32-year-old female. She has had previous vision problems and pedal edema but was being treated by local village doctors when finally, she presented to us with de-compensated renal failure. The other disorders that can present with retinal dystrophies are Usher syndrome, Kearns–Sayre syndrome, Bassen–Kornzweig syndrome, and Refsum syndrome. Alström syndrome (AS) is a very rare autosomal recessive genetic disorder characterized by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss, and dilated cardiomyopathy. Developmental delay is seen in almost half of people with AS. However, our patient had a normal 2D Echo and no type 2 diabetes mellitus. Other syndromes that could be confused with Bardet–Beidl syndrome are McKusick–Kaufmann syndrome, Joubert syndrome, Jeune syndrome, Sensenbrenner syndrome (cranioectodermal dysplasia), and Senior–Loken Syndrome.
The term Bardet–Beidl syndrome was coined by Georges Berdet and Arthur Beidl. BBS is inherited in autosomal recessive fashion and hence more common in a person born of consanguineous marriage such as in our case. A pedigree chart of inheritance has been shown in Figure 3.

- Pedigree chart of the Patient's Family
BBS is genetically heterogeneous and has four mapped loci, namely, BBS1 (11q13),[6] BBS2 (16q22),[7] BBS3 (3p13),[8] and BBS4 (15q21).[9] Later on, eight more genes were coded. Presently, 12 genes (BBS1 to BBS12) for the disease have been cloned.[10] The proteins associated with BBS are components of the centrosome that play a major role in ciliary transport; hence, the disease has been classified under “ciliopathies.”[11] The patient we studied had a BBS 9 variant (MIM#615896) with a missense mutation in c.711 G>T (p.W237C) at Exon 8 of Chromosome 7 (Chr7:33312632). The missense mutation we spotted here has not been reported previously as pathogenic or as benign and is a novel variant. For this reason, it has been classified as of 'Uncertain Significance'. BBS 9 is caused by homozygous or compound heterozygous mutation in the PTHB1 gene.[12]
The cardinal features of Bardet–Biedl syndrome are central obesity [Figure 1c], post-axial polydactyly [Figure 2a], rod-cone dystrophy (also called atypical retinitis pigmentosa) [Figure 2b], mental retardation, hypogonadism, and renal dysfunction.[1314] In males, hypogonadism was very common. In females, the mean age of menarche was 13.8 years.[15] Following the onset of menstruation, irregular cycles were reported for the majority of females.
The primary concern in our patient was end-stage renal failure. We decided to put her on maintenance hemodialysis twice/week. A arterio-venous fistula was created in her right arm for further dialysis. The management of chronic kidney disease in BBS is the same as in other conditions, and all three methods of renal replacement therapy, that is, hemodialysis, chronic peritoneal dialysis, and renal transplantation, can be used.[3] A low-calorie and low-protein diet help in obesity control and may slow the progression of renal failure in patients with BBS.[16]
We are reporting this case for its rarity and the presence of a novel genetic variant of an unidentified significance as per genome mapping. BBS is often not diagnosed at all or diagnosed late until end-stage renal failure sets in. No definitive treatment exists for BBS. However, early diagnosis not only helps in actively seeking and identifying co-morbidities but also helps in management of co-morbidities, thus retarding the progression to CKD and improving the quality of life.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We express our sincere gratitude to the authors who participated in our study. All authors have equal contribution in the work. We are also thankful to the patient and her family members for their cooperation during their hospital stay. No funding by the institute or external source was required for the following study.
References
- A case of Bardet-Biedl syndrome complicated with intracranial hypertension in a Japanese child. Brain Dev. 2014;36:721-4.
- [Google Scholar]
- The syndrome of Laurence-Moon-Bardet-Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci. 1969;9:479-513.
- [Google Scholar]
- Renal transplant in a child with Bardet-Biedl syndrome: A rare cause of end-stage renal disease. Indian J Nephrol. 2009;19:112-4.
- [Google Scholar]
- Age-related cognitive decline and vision impairment affecting the detection of dementia syndrome in old age. Br J Psychiatry. 1997;171:449-51.
- [Google Scholar]
- Chapter 2: Definition, identification, and prediction of CKD progression. Kidney Int. 2013;3(l):63-72.
- [Google Scholar]
- Bardet-Biedl syndrome is linked to DNA markers on chromosome 11q and is genetically heterogeneous. Nat Genet. 1994;7:108-12.
- [Google Scholar]
- Linkage of Bardet-Biedl syndrome to chromosome 16q and evidence for non-allelic genetic heterogeneity. Nat Genet. 1993;5:392-6.
- [Google Scholar]
- Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping. Hum Mol Genet. 1993;3:1331-5.
- [Google Scholar]
- Use of a DNA pooling strategy to identify a human obesity syndrome locus on chromosome 15. Hum Mol Genet. 1995;4:9-13.
- [Google Scholar]
- Bardet-Biedl syndrome: A rare case report from North India. Indian J Dermatol Venereol Leprol. 2012;78:228.
- [Google Scholar]
- A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201-13.
- [Google Scholar]
- The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989;321:1002-9.
- [Google Scholar]
- The spectrum of renal disease in Laurence-Moon-Biedl syndrome. N Engl J Med. 1988;319:615-8.
- [Google Scholar]
- New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J Med Genet. 1999;36:437-46.
- [Google Scholar]
- Obesity control and low protein diet preserve or even improve renal functions in Bardet-Biedl syndrome: A report of two cases. Case Reports Clin Pract Rev. 2011;17:2009-11.
- [Google Scholar]




