

Translate this page into:
C1q Nephropathy in a Patient of Neurofibromatosis Type 1: A Rare Case Report
Address for correspondence: Dr. U. T. Varyani, Department of Nephrology and Transplantation Medicine, G.R. Doshi and K.M. Mehta Institute of Kidney Diseases and Research Centre - Dr. H. L. Trivedi Institute of Transplantation Sciences, Civil Hospital Campus, Asarwa, Ahmedabad - 380 016, Gujarat, India. E-mail: umesh08varyani@gmail.com
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
C1q nephropathy is a rare glomerular disease defined by the presence of characteristic mesangial dominant or codominant C1q deposition on immunofluorescence microscopy. Neurofibromatosis type 1 (NF-1) is an autosomal dominant syndrome caused by a mutation of a gene located on chromosomal segment 17q11.2. Nephrotic syndrome has rarely been reported in patients of NF-1, and the relation of NF-1 with nephrotic syndrome is unclear. Here, we present a rare case of C1q nephropathy in a patient of NF-1.
Keywords
C1q nephropathy
focal segmental glomerulosclerosis
neurofibromatosis

Introduction
C1q nephropathy is defined as the presence of mesangial C1q staining either dominant or codominant with the absence of clinical or serological findings of systemic lupus erythematosus (SLE). It was first described by Jennette and Hipp in 1985.[1] It has varied light microscopic features including minimal change nephropathy, focal segmental glomerulosclerosis (FSGS), and crescentic glomerulopathy. It may or may not present with nephrotic syndrome.[2] Neurofibromatosis type 1 (NF-1) is the most common neurocutaneous disorder inherited in an autosomal dominant fashion due to a mutation in NF-1 gene. The NF-1 gene is located at chromosome 17q11.2 and encodes protein neurofibromin.[3] Association of NF-1 with glomerulopathy is rare. To our knowledge, this is the first case report of C1q nephropathy described in association with NF-1.
Case Report
A 51-year-old married male presented with complaints of swelling over lower limbs, face, and abdomen along with frothy urine for last 1 month. There was no history of preceding fever, sore throat, joint pain, rash, or hematuria. There was no past history of similar complaints, hypertension, diabetes, chronic analgesic or other drug abuse, blood transfusion, or any high-risk sexual behavior.
On admission, he was conscious, cooperative, and oriented to time, place, and person. He was afebrile; had a pulse rate of 82 beats/min, regular, normal volume; and had blood pressure of 126/82 mmHg with a respiratory rate of 18 breaths/min. There was the presence of facial puffiness and pitting pedal edema. On per abdomen examination, there was generalized abdominal distention with fullness of flanks, and on percussion, there was shifting dullness confirming the presence of ascites. His respiratory and cardiovascular examination was unremarkable. He had multiple purplish, well-circumscribed, and pedunculated skin lesions over face, chest, abdomen, and back. [Figure 1b] Also, on the trunk was presence of many café-au-lait spots of more than 1.5 cm in diameter.[Figure 1a] Ophthalmological examination revealed two Lisch nodules in iris in the left eye and three in the right eye. There was a history of similar skin lesions in patient's father and paternal grandfather although no history of renal disease was present. The diagnosis of NF-1 was made as per the National Institute of Health Criteria [Table 1].

- (a) Presence of multiple café-au-lait macules and (b) shows the presence of neurofibromas on back
| The patient should have two or more of the following |
| Six or more café-au-lait spots or hyperpigmented macules >5 mm in diameter in prepubertal children and 15 mm in postpubertal |
| Axillary or inguinal freckles (>2 freckles) |
| Two or more typical neurofibromas or one plexiform neurofibroma |
| Optic nerve glioma |
| Two or more Lisch nodules |
| Sphenoid dysplasia or typical long-bone abnormalities such as pseudarthrosis |
| First-degree relative with NF1 |
NF1: Neurofibromatosis 1
His laboratory investigations showed hemoglobin 13.7 g/dl, white blood cell (WBC) 10,500/mm3 and platelets 3.61 lakhs, creatinine 0.98 mg/dl, urea 48 mg/dl, sodium 135.3 mEq/l, potassium, 4.81 mEq/l, calcium (Ca) 6.1 mg/dl with corrected Ca of 7.6 mg/dl, proteins 4.5 g/dl, albumin 2.1 g/dl, globulins 2.40 g/dl, total cholesterol 371 mg/dl, random blood glucose 87 mg/dl, serum antinuclear and antidouble-stranded DNA antibodies by ELISA were negative, C3 148 mg/dl (N 90–207 mg/dl), and C4 47.2 mg/dl (N 17.4–52.2 mg/dl). ELISA for HIV and hepatitis B and C was nonreactive. Urine routine and microscopic analysis revealed the presence of proteinuria 4+, red blood cell 40–50/HPF, WBC nil, and no cellular casts. Twenty-four-hour urinary proteins were 5.1 g.
Ultrasound abdomen examination revealed the presence of ascites with fused kidneys present on the right side with preserved corticomedullary differentiation.
A percutaneous kidney biopsy was performed under ultrasound guidance. Kidney biopsy pathological findings revealed the presence of 12 glomeruli with two glomeruli showing segmental mesangial sclerosis along with occasional synechiae formation with normal capillary membrane thickness and unremarkable Bowman's capsule. There were mild interstitial mononuclear cellular infiltration and mild tubular degeneration. [Figure 2a and b] Immunofluorescence examination showed fine granular C1q (+3/+4) and IgG (+2) positivity across 70%–80% of the mesangial regions of all glomeruli with negative IgA, IgM, and C3. [Figure 2c and d] Therefore, a diagnosis of C1q nephropathy was made.
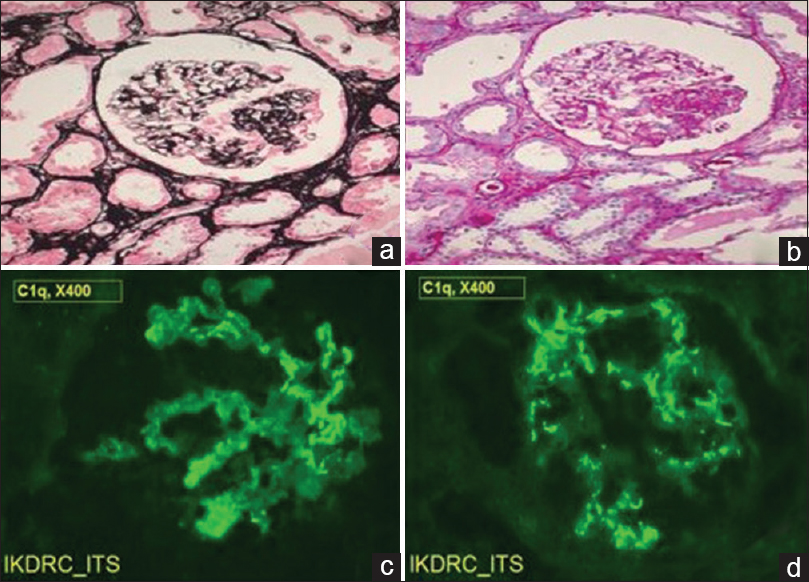
- (a) (Jones methenamine silver, ×100) and (b) (Periodic acid–Schiff, ×100) showing focal segmental mesangial sclerosis with synechiae formation. (c and d) (Immunofluorescence, ×400) shows fine granular staining with antihuman C1q in mesangial regions of glomeruli
The patient was started on treatment with prednisolone 1 mg/kg/day (50 mg) and telmisartan 20 mg once a day. Patient achieved complete remission after 14 weeks of steroid therapy with complete resolution of facial and pedal edema with 24 h urinary protein reduced to 280 mg and hence steroid dose was subsequently tapered. After 5 months of treatment, the patient is still in complete remission and at present is on 20 mg of prednisolone.
Discussion
NF-1 is the most common neurocutaneous disorder with an autosomal dominant inheritance with an incidence of 1 in 3500 live births.[3] It has varied clinical presentations including neurofibromas, café-au lait spots, axillary or inguinal freckling, Lisch nodules, and characteristic bony lesions (pseudarthrosis and sphenoid wing hypoplasia).[4] Our patient had the presence of cutaneous neurofibromas, more than 6 café-u lait spots more than 1.5 cm in diameter, Lisch nodules in iris, axillary freckling, and positive family history of similar skin lesions. Therefore, diagnosis of NF-1 was made as per the National Institute of Health criteria [Table 1]. NF-1 is caused by mutations in gene encoding neurofibromin located at chromosome 17q11.2.[4] NF-1 can involve the kidneys with the most common presentation being renal artery stenosis.[56] Glomerulopathy is rare in NF-1 with only a few cases reported. Membranous glomerulopathy is the most common glomerulopathy reported in association with NF-1. The others include FSGS, IgA nephropathy, and MCN.[789101112] Our patient had presented with nephrotic syndrome and renal biopsy on light microcopy was suggestive of FSGS. IF microscopy showed the presence of fine granular (+3/+4) fluorescence across 70%–80% of the mesangial regions of all glomeruli on staining with anti-human C1q and (+2) with IgG antisera.
C1q nephropathy is a rare glomerular disease characterized by the presence of mesangial C1q staining either dominant or codominant with the absence of clinical or serological findings of SLE. The pathogenesis of C1q nephropathy is unclear. C1 is the first component of classical pathway of complement system. Classical pathway activation involves binding of C1q to Fc region of IgG and IgM containing immune complexes. C1q receptors are present in mesangial cells. It has been proposed that C1q binds to immunoglobulins trapped nonspecifically in mesangium in course of glomerular proteinuria.[213]
C1q nephropathy has heterogeneous light microscopic features. Vizjak et al. studied 82 kidney biopsies from 28 children and 54 adults. IF microscopy showed dominant or codominant staining for C1q in the mesangium, Light microscopy revealed no lesions (n = 27), focal segmental glomerulosclerosis (FSGS; n = 11), proliferative glomerulonephritis (n = 20), or various other lesions (n = 14). All patients with FSGS presented with nephrotic syndrome.[14] Our patient had also presented with nephrotic syndrome and had FSGS on LM.
The treatment of C1q nephropathy depends on underlying light microscopic lesion. The mainstay of treatment is glucocorticoids. Cyclophosphamide, cyclosporine, tacrolimus, or mycophenolate mofetil have been used in patients unresponsive to steroids. Minimal change disease has a more favorable response to therapy as compared to FSGS. Vizjak et al. followed up 53 patients with C1q nephropathy; 76.9% of the minimal change-like group but only one-third (33.3%) of the FSGS group were in complete remission after 4 months to 21 years, and four patients had partial remission after 4 months to 3 years.[15]
There were no clinical features of SLE, and serological markers for lupus were absent in our patient (ANA and anti-dsDNA). Hence, the diagnosis of C1q nephropathy was made, and in view of nephrotic syndrome, patient was started on prednisolone 1 mg/kg, to which patient responded well and achieved complete remission in 14 weeks.
The activation of mitogen-activated protein kinase and mammalian target of rapamycin signaling pathways due to deficient neurofibromin as a mechanism for the development of FSGS and nephrotic range proteinuria in NF1 were proposed by Farsad et al.[16]
Conclusion
NF-1 has been rarely associated with various glomerular diseases. To our knowledge, this is the first case of C1q nephropathy in association with NF-1, but their independent occurrence could not be determined.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The author acknowledges our chief librarian Ms. J Suthar for literature search. Authors are also thankful to the junior resident doctors for their help.
References
- C1q nephropathy: A distinct pathologic entity usually causing nephrotic syndrome. Am J Kidney Dis. 1985;6:103-10.
- [Google Scholar]
- Diverse clinical and histology presentation in C1q nephropathy. Nephrourol Mon. 2013;5:787-91.
- [Google Scholar]
- Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol. 2006;13:2-7.
- [Google Scholar]
- A case of membranous glomerulonephritis associated with Recklinghausen's neurofibromatosis. Clin Nephrol. 1996;45:271-2.
- [Google Scholar]
- Focal and segmental glomerulosclerosis and von Recklinghausen's neurofibromatosis: Coincidental or associated? Saudi J Kidney Dis Transpl. 2008;19:453-4.
- [Google Scholar]
- Focal segmental glomerular sclerosis in a patient with neurofibromatosis type I. Am J Kidney Dis. 2006;47:e17-9.
- [Google Scholar]
- Parent and child cases of IgA nephropathy associated with von Recklinghausen's disease. Nephron. 1997;75:113-4.
- [Google Scholar]
- Von Recklinghausen's neurofibromatosis associated with membranous glomerulonephritis. Saudi Med J. 2006;27:534-5.
- [Google Scholar]
- C1q nephropathy: A variant of focal segmental glomerulosclerosis. Kidney Int. 2003;64:1232-40.
- [Google Scholar]
- Minimal change disease in a patient with neurofibromatosis type I. Acta Nephrol. 2012;26:32-234.
- [Google Scholar]
- Pathology, clinical presentations, and outcomes of C1q nephropathy. J Am Soc Nephrol. 2008;19:2237-44.
- [Google Scholar]
- Focal segmental glomerulosclerosis in association with neurofibromatosis type 1: A case report and proposed molecular pathways. Clin Kidney J. 2013;6:208-10.
- [Google Scholar]




