

Translate this page into:
Catastrophic antiphospholipid antibody syndrome in a child with thrombotic microangiopathy
Address for correspondence: Dr. Narayan Prasad, Department of Nephrology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, Pin - 226 014, India. E-mail: narayan@sgpgi.ac.in
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Thrombotic microangiopathic hemolytic anemia (TMHA) is not uncommon in clinical nephrology practice while antiphospholipid syndrome (APS) is uncommon. Although less than 1% of patients with APS develop catastrophic APS (CAPS), its potential lethal outcome because of thrombosis in multiple organs and subsequent multiorgan failure emphasizes its importance in nephrology practice. Here is a case of catastrophic APS in a 7-year-old girl, who presented to us with TMHA associated with antiphospholipid antibodies and subsequently died because of CAPS.
Keywords
Catastrophic antiphospholipid syndrome
renal failure
thrombotic microangiopathic hemolytic anemia

Introduction
Thrombotic microangiopathic hemolytic anemia (TMHA) is not uncommon in nephrology practice, while antiphospholipid syndrome (APS) is uncommon. Although less than 1% of patients with APS develop the catastrophic variant, its potentially lethal outcome with multiorgan failure emphasizes its importance in clinical nephrology today. TMHA associated with antiphospholipid (aPL) antibodies is uncommon and has been reported in only 7% of patients in a series of 1,000 patients with APS.[1] In a study of 47 episodes of TMHA associated with APS, only 23% had clinical presentation of catastrophic APS (CAPS).[2] Patients with CAPS (currently also known as Asherson's syndrome)[3] show clinical evidence of multiple organ involvement developing over a very short period of time; histopathological evidence of multiple small vessel occlusions; and aPL antibodies, usually in high titers. TMHA is characterized by thrombocytopenia, microangiopathic hemolytic anemia accompanied by a negative Coombs’ test result, fever, neurological symptoms, and kidney involvement.[2] Durand et al. have pointed out the relationship of TMHA with the presence of aPL antibodies.[3] Espinosa et al. have shown that TMHA might be a manifestation of APS.[2] There may be an overlap in clinical presentation of TMHA and CAPS.[4] Moreover, in the CAPS registry, only few cases of CAPS in children were reported. Hence, we report a rare case of APS in a 7-year-old girl who presented with TMHA and subsequently developed CAPS.
Case Report
A 7-year-old girl presented to the nephrology clinic with complaints of fever for 3 days followed by jaundice, cola colored urine, vomiting, and swelling of body. On examination, she had pulse of 86/min, blood pressure (BP) 110/80 mm Hg, pallor, jaundice, and edema. She had received three units of blood transfusion before this hospitalization. She had cola color urine before blood transfusion. As the patient was from malaria endemic zone, a possibility of complicated malaria causing acute kidney injury was considered. She had been treated with antimalarial artemether. Her hemogram revealed hemoglobin of 9.9 g/dl, total leukocyte count 12,000/mm3 (polymorph 85%, lymphocyte 12%, eosinophil 2%, and monocytes 1%) platelet count 23,000/mm3, and reticulocyte count 6.6%. Her peripheral blood picture revealed multiple fragmented red cells and polychromasia. Her thick blood smear for malaria parasite showed negative result. Her biochemical parameters revealed blood urea nitrogen 110 mg/dl, serum creatinine 6.6 mg/dl, Na+ 136 mmol/L, K+ 3.5 mmol/L, protein 6.6 gm/dl, albumin 3.7 gm/dl, total cholesterol 176 mg/dl, triglyceride 160 gm/dl, total bilirubin 5 mg/dl with indirect fraction 3.3 mg/dl, lactate dehydrogenase 1342 IU/L (normal 85-450 IU/L), and negative direct and indirect Coomb's test. Her coagulation parameters, viz. prothrombin time (PT) and activated partial thromboplastin time (APTT) yielded normal results. Her ultrasonogram of the abdomen revealed normal-sized kidneys with increased echogenicity with normal corticomedullary differentiation, and evidences of cholecystitis with granular sludge in the gall bladder. TMHA was considered in the event of history of febrile illness, cola colored urine, jaundice, Coomb's negative hemolytic anemia, thrombocytopenia, reticulocytosis, and renal failure. Kidney biopsy was deferred in view of low platelet count at that time. She had been treated with two sessions of heparin-free intermittent hemodialysis and plasma infusion. She became afebrile for two days; urine became clear, serum creatinine decreased to 2.8 mg/dL, and also improved symptomatically.
On the fifth day of admission, she again developed fever with cola colored urine, one episode of generalized tonic-clonic seizures, and right-sided hemiplegia. Her immediate computed tomography (CT) scan of the cranium was normal. However, the magnetic resonance imaging of her brain revealed multiple infarcts of varying sizes [Figure 1a], with the largest one in the left frontal-parietal region; angiography of cerebral vessels revealed thrombosis in the common carotid and cerebral vessels [Figure 1b, c]. Her repeat PT was normal and APTT was prolonged (test 45 sec, control 25 sec). The flow cytometry for paroxysmal nocturnal hemoglobinuria (PNH) was negative. She had normal C-ANCA, P-ANCA, complements level (C3, C4), negative antinuclear antibody, anti-Rho, anti-La, and anti dsDNA titer. Her anticardiolipin antibody [IgG] was positive and lupus anticoagulant activity was found to be positive in high titer (1:64). A clinical diagnosis of catastrophic antiphospholipid syndrome was confirmed and she had been treated with heparin, dexamethasone, five sessions of plasma exchanges, and intermittent hemodialysis every other day. Intravenous immunoglobulin could not be given due to its high cost.

- (a) Magnetic resonance imaging (T1W) showing acute infarct in the left frontal, temporal, and parietal region, right centrum semiovale, and periventricular region. (b) Magnetic resonance angiography with coronal MIP projection of circle of Willis showing paucity of distal branches (arrows) of the left Middle Cerebral Artery (MCA) compared to the right side. (c) Magnetic resonance angiography (3D MIP reconstruction TOF) showing filling defects in basilar artery (thin arrow), focal narrowing of the right internal carotid artery (curved arrow), and M-2 segment of the right MCA (thick arrow)
The patient developed refractory status epilepticus despite antiepileptic drugs such as phenytoin, sodium valproate, and levetiracetam. She was put on mechanical ventilation due to refractory status epilepticus and impending respiratory arrest. She developed tachyarrhythmia and echocardiography revealed hypokinesia of the apical segment of heart, suggestive of coronary involvement. She developed ventilator associated pneumonia, sepsis, and died of cardiac arrest due to refractory ventricular fibrillation. Postmortem kidney biopsy was done with informed consent of parents. Histopathology was suggestive of thrombotic microangiopathy [Figure 2].
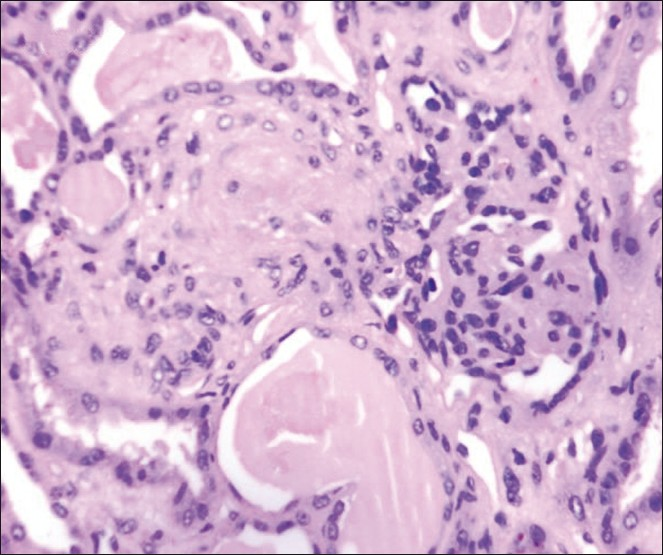
- Histopathologic examination of the renal biopsy showing ischemic wrinkling of glomerular capillary basement membrane with endothelial cell swelling and obliteration of capillary lumina, and associated arteriole with fibrinoid necrosis of the wall and luminal obliteration (M, ×200)
Discussion
This girl died of CAPS, an uncommon and lethal form of APS. CAPS is characterized by multifocal rapidly progressive thrombosis that leads to multiorgan failure.[5] This patient had all evidences of TMHA at presentation. The differential diagnoses of TMHA in the pediatric patient include sporadic hemolytic uremic syndrome (HUS) and/or thrombotic thrombocytopenic purpura (TTP). The triad of thrombocytopenia, microangiopathic hemolytic anemia, and renal failure satisfied the diagnostic criteria of HUS. Moreover, the presence of fever and neurological involvement in the subsequent course satisfied the pentad criteria of TTP. The diagnosis of CAPS was only confirmed once the patient had developed hemiparesis, seizure, and magnetic resonance angiography revealed diffuse thrombosis of the major carotid and cerebral vasculature and subsequently the presence of aPL antibodies.
Malarial acute renal failure (ARF) was ruled out with the absence of malarial antigen and malaria parasite on smear. With controlled blood pressure and normal fundus examination, malignant hypertension was unlikely. Other differential diagnoses of TMHA such as cancer, immunosuppressive treatment, systemic sclerosis, undifferentiated connective tissue disorder, and human immunodeficiency virus infection were ruled out due to the absence of circumstantial evidences of these diseases.
Negative antinuclear antibody (ANA), normal anti-dsDNA titer and normal complement ruled out systemic lupus erythematosus (SLE) and APS secondary to SLE in this patient. SLE was the first autoimmune disease in which the association of TMHA with aPL antibodies was recognized. However, renal lesions of TMHA might develop during the course of TTP or HUS or be associated with APS, regardless of the underlying type of lupus glomerulopathy.
The distinction between catastrophic APS and TTP/HUS was of vital importance. Anticoagulant treatment is considered as the first-line therapeutic approach in the former, while it does not benefit the latter.[6] Infection is reported to be a contributing precipitating factor in CAPS. Salmonella, upper respiratory infection, and others have been well documented to precede CAPS.[7] We could not isolate any infection probably due to prior antibiotic treatment before presentation to this hospital; however, fever and leukocytosis at presentation may be a suggestive factor of infection precipitating CAPS.
There have been more than 280 documented cases of CAPS since 1992.[8] Probably, more non-reported cases might exist worldwide, with mortality rate of over 50% even in adequately treated patients, and death is usually caused by multiorgan failure. Despite being recognized as an important cause of vascular thrombosis in the pediatric population, only few CAPS has been reported in children.[9–12] A case of CAPS has been reported in a child with pediatric SLE,[9] and in another child with myelomonocytic leukemia.[10] Park et al. have reported a case of CAPS in a 7-year-old child who survived without recurrence for 3 years on follow-up.[12] Tsirpanlis et al. have reported a case in a 14-year-old child who also survived despite multiorgan failure at presentation.[13] Our patient died despite prompt diagnosis because of a large cerebral infarction, refractory seizure that needed ventilatory support and subsequently developed ventilator-associated pneumonia. Our patient was unique, one of the youngest and a rare case of CAPS in pediatric population, who developed CAPS in the subsequent course of TMHA. However, the patient did not survive long enough for repeat testing of antibodies. Other cases of CAPS in pediatric population did not have this much evident TMHA. In a series of 47 episodes of TMHA associated with APS, 26% had clinical presentation of HUS, 23% with CAPS, 13% with malignant hypertension, 13% with TTP, and 19% had pregnancy-associated TMHA (2). Although the pathogenesis of CAPS remains unclear, the role of endothelial cell activation is suggested. It is uncertain whether aPL antibodies are directly involved in this. Endothelial cell injury is a common denominator in TMHA of diverse etiologies, but the inciting factors vary greatly. In TTP, it is secondary to deficient metalloprotease, proteolytic enzyme ADAMTS13 in plasma or, an antibody against this enzyme, while in classic HUS, the Shiga toxin triggers endothelial injury.
The therapeutic connotation of CAPS is that it may be corrected with a combination of anticoagulation plus steroids plus attempts at achieving a prompt reduction in aPL titer with plasma exchange and/or intravenous immunoglobulin. Our patient was administered heparin, dexamethasone, and plasma exchange.
In conclusion, CAPS should be considered as differential diagnoses of TMHA in pediatric patient population. We need to bear in mind that APS could be a cause of thromboses, affecting multiple organs in children with multi-organ involvement with positive APLA and lupus anticoagulant activity.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Antiphospholipid syndrome: Clinical and immunologic manifestations an patterns of disease expression in a cohort of 1000 patients. Arthritis Rheum. 2002;46:1019-27.
- [Google Scholar]
- Thrombotic microangiopathic hemolytic anemia and antiphospholipid antibodies. Ann Rheum Dis. 2004;63:730-6.
- [Google Scholar]
- Thrombotic microangiopathy and the antiphospholipid antibody syndrome. J Rheumatol. 1991;18:1916-8.
- [Google Scholar]
- Thrombotic microangiopathic haemolytic anemia (thrombotic microangiopathy) Br Med J. 1952;2:897-903.
- [Google Scholar]
- Catastrophic antiphospholipid syndrome: Clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
- [Google Scholar]
- Catastrophic antiphospholipid syndrome. Where do we stand? Arthritis Rheum. 2003;48:3320-7.
- [Google Scholar]
- Thrombotic microangiopathy, haemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Kidney Int. 2001;60:831-46.
- [Google Scholar]
- Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of a series of 280 patients from the “CAPS Registry”. J Autoimmun. 2009;32:240-5.
- [Google Scholar]
- Catastrophic antiphospholipid antibody syndrome in pediatric systemic lupus erythematosus. J Rheumatol. 1997;24:389-92.
- [Google Scholar]
- Childhood acute myelomonocytic leukemia (AML-M4) presenting as catastrophic antiphospholipid antibody syndrome. J Pediatr Hematol Oncol. 2004;26:327-30.
- [Google Scholar]
- Catastrophic antiphospholipid syndrome in a 7-year-old girl. Clin Rheumatol. 2007;26:1011-3.
- [Google Scholar]
- Catastrophic antiphospholipid syndrome in a 14-year-old child. Pediatr Nephrol. 2005;20:519-21.
- [Google Scholar]




