

Translate this page into:
Diuretic loading test and use of Bartter's Normogram in diagnosing a case of Gitelman's syndrome: Relook into pathophysiology
Address for correspondence: Dr. Rudra Goswami, Abhyudoy Housing, Flat - 18/14, Kolkata, India. E-mail: rudra.goswami@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Gitelman's syndrome is a rare autosomal recessive, renal tubular disorder, characterized by chronic hypokalemia, hypomagnesemia, metabolic alkalosis, hypocalciuria, and normal blood pressure. Patients usually present at a later age with episodic mild muscle weakness. Unexplained hypokalemia arouses suspicion. The diuretic loading test with furosemide and thiazide and the use of Bartter's normogram provides a practical and simple tool in comparison to the complex and costly genetic analysis, to confirm the diagnosis. Here we report a case of Gitelman's syndrome to show the utility of these simple techniques to explain the pathophysiology of the disease, as well as to localize the site of the renal tubular defect, to confirm the diagnosis.
Keywords
Bartter's normogram
diuretic loading test
Gitelman's syndrome

Introduction
Gitelman's syndrome (GS) is a rare autosomal recessive, renal tubular disorder characterized by chronic hypokalemia, hypomagnesemia, metabolic alkalosis, hypocalciuria, modest hyperreninemia, minimal-to-absent volume depletion, and a low normal blood pressure, with a normal glomerular filtration rate. This disorder is linked to the gene encoding the thiazide sensitive Na+-Cl–co-transporter (NCC) located on chromosome 16q[1–3] (without affecting the furosemide-sensitive carrier). This report presents an affected 52-year-old female, with emphasis on distal nephron functional studies with furosemide and hydrochlorothiazide and reviews GS.
Case Report
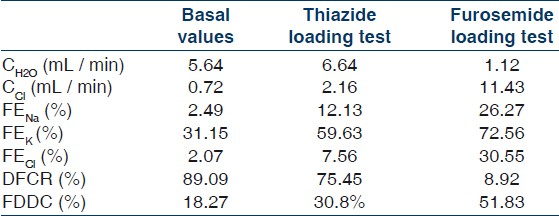
The 52-year-old female, a housewife, presented with a history of an unknown insect bite in the medial part of her left thigh, three weeks ago, followed by development of a deep crater like ulcer with gradual development of fever in an unremitting course. After completion of systemic antibiotic therapy and proper surgical debridement of her thigh lesion the patient was seen to be in a haggard condition lying in a listless posture. On detailed questioning she reported bilateral proximal upper and lower limb muscle weakness, which was objectively confirmed. On further probing several such episodes were revealed, which used to precipitate during exertion. Throughout her course in the hospital she was normotensive (110/70 mmHg ± 7/5 mmHg), with a normal renal function (serum urea – 13 mg/dL and creatinine – 0.72 mg/dL). On evaluating the muscle weakness, persistent hypokalemia was noticed, with a potassium level of 1.31 (±0.75) mmol/L. An arterial blood gas showed metabolic alkalosis and a detailed biochemical examination showed hypocalcemia and hypomagnesemia. Both her plasma rennin activity and plasma aldosterone level were modestly elevated [Table 1].

To evaluate the hypokalemia and metabolic alkalosis, we excluded surreptitious diuretic use, extracellular volume contraction (diarrhea or vomiting) or a post-hypercapnic state. Urinary electrolytes were measured, which revealed urine Cl- above 20 mEq / day, which excluded a chloride-responsive cause of metabolic alkalosis, and a urine K+ above 30 mEq / day, which was consistent with a primary tubular disorder. A family history of Bartter's syndrome was absent and a provisional diagnosis of Gitelman's syndrome was made. Urine calcium estimation was made, which showed hypocalciuria. An ultrasonographic examination of the abdomen did not reveal any evidence of nephrocalcinosis, and an electrocardiogram (ECG) and stress echocardiography were within normal limits.
To assess the tubular function, diuretic challenge tests were contemplated and informed consent was obtained from the patient. She was interrupted from any pharmacological therapy for 10 days before the studies and allowed an unrestricted diet. The renal clearance studies were performed using the protocol described earlier.[145] After overnight fasting, the patient drank water (20 ml / kg of body weight) over 20 minutes and received intravenous infusion of 0.45% saline at a rate of 40 ml / hour. Urine output was measured every 20 minutes followed by additional water intake (urine volume plus 20 ml / 20 minutes).When urine flow reached a maximum level, samples of blood and urine were obtained, to calculate the basal values of the parameters described a little later in the text. For thiazide loading test, 100 mg of hydrochlorothiazide was orally administered to the patients. Water loading was carried out. For the furosemide loading test, 40 mg of furosemide was given intravenously to the patients. In both tests, the clearance data were obtained when the urine flow was maximum. Furosemide and Hydrocholorthiazide tests were carried out seven days apart.
The parameters used were:
Solute-free water clearance (CH2O) = V × (1 – Uosm/Posm), where V = urine flow rate in mL / min, Uosm = urinary osmolality, Posm = plasma osmolality.
Chloride clearance (CCl) = V × UCl/PCl, where UCl = urinary chloride concentration, PCl = plasma chloride concentration.
Distal fractional chloride reabsorption (DFCR) = CH2O/ (CH2O + CCl)
Fractional distal delivery of chloride (FDDC) = (CH2O + CCl) / Creatinine clearance
Fractional excretion of a solute [X] (FEX) = 100 × (UX/PX) × (Pcr/ Ucr), where, Pcr = plasma creatinine concentration, Ucr = urinary creatinine concentration, X was taken as Na (sodium), K (potassium), and Cl (chloride).
The basal values showed a markedly high value of FEK of 31.15% (normal 4 – 16% with a mean of 8%[5]). The furosemide challenge test showed a significantly increased FDDC and a significantly decreased DFCR. Neither of these parameters was markedly altered by Thiazide. Furosemide loading resulted in a marked increase in FENa, FEK, and FECl, with reduced free water clearance and chloride clearance, while Thiazide loading did not significantly alter the free water clearance or chloride clearance, but did result in an increased fractional excretion of solutes, but to a lesser extent than that caused by furosemide [Table 2].
Discussion
Gitelman's syndrome (GS), also known as a ‘hypocalciuric variant’ of Bartter's syndrome (BS) or as ‘familial hypokalemia-hypomagnesemia’, is an autosomal recessive inherited disorder, first described by Gitelman et al., in 1966.[6]
GS is caused by inactivating mutations in the gene for the thiazide sensitive NCC (thiazide-sensitive NCC) of the distal convoluted tubule, called SLC12A3.[278] It should be suspected in patients with hypokalemia, metabolic alkalosis, and hypocalciuria. Patients with Gitelman's syndrome are usually normotensive, which differentiate them from patients with Liddle's syndrome. Hypomagnesemia, although an important feature of the typical GS, may be absent in some patients. Patients with GS present in adult life, differentiating it from most forms of Bartter's syndrome. Most patients with GS are asymptomatic or complain of mild intermittent cramps or muscle weakness,[4] with a few developing chondrocalcinosis, with increased bone mineral density, choroidal calcification or rarely prolongation QT interval, with development of fatal cardiac arrhythmia.[9]
In our patient, the diagnosis of GS was made after the patient was found lying listlessly, and had muscle weakness. Probing revealed history of previous episodes of intermittent muscle weakness. As primary hypokalemic periodic paralysis rarely presents after the age of 25 years,[10] a diagnosis of secondary hypokalemic periodic paralysis was entertained. The classical presentation of hypokalemia, hypomagnesemia, hypocalcicuria, a normal blood pressure, modestly raised plasma rennin, and aldosterone activity clinches the diagnosis of Gitelman's syndrome. Our patient did not share any of the more sinister complications listed here.[11]
In our study, thiazide induced blunted natriuresis and chloruresis as compared to the markedly increased values obtained with furosemide. The CCl and DFCR showed an abrupt change after furosemide administration, while thiazide had little effect on our patient. The most likely explanation for this data is the decreased diuretic effect of thiazide in GS patients, owing to the genetic defect of GS resulting in reduced NaCl reabsorption via the thiazide sensitive Na+/Cl–symporter of the distal convoluted tubule.
The relationship between FDDC and DFCR is shown in Figure 1, with data borrowed from Gill and Bartter.[10] Our patients’ basal values were in between the surreptitious vomiting and Bartter's syndrome (BS) groups, but closer to the former. Thiazide loading dampened the value in our patient closer to the BS group.
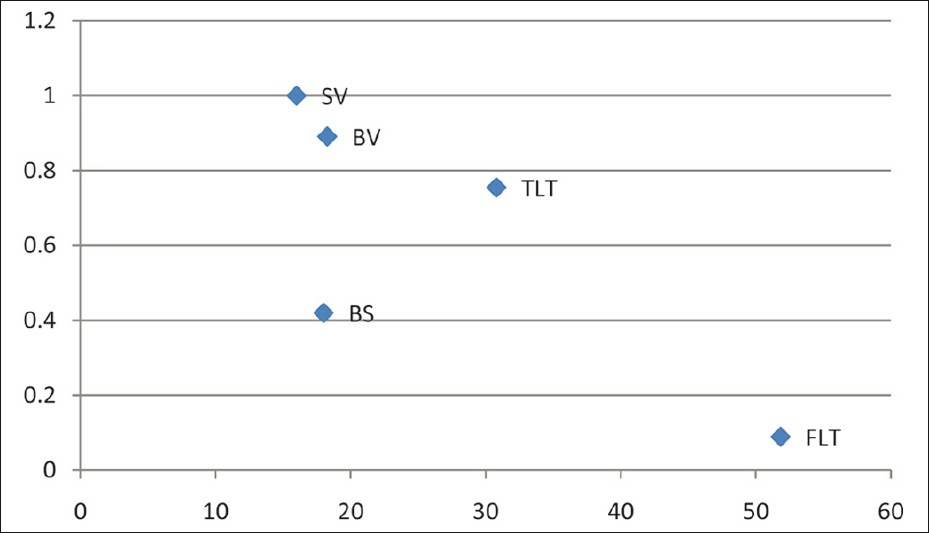
- FDDC (x – axis) versus DFCR (y – axis) (SV = Surreptious vomiting; BS = Bartter's syndrome; BV = Basal value; TLT = Thiazide loading test; FLT = Furosemide loading test)
Similar results were also obtained by previous workers.[134] In agreement with the previous studies we believe this differential response owes it origin to the differential placement of receptors for the different diuretics. As furosemide acts in the loop of Henle that is anatomically proximal to the distal convoluted tubule (DCT), which is the primary site of the receptor defect in GS, administration of furosemide results in increased distal delivery of the chloride, as reflected by the increased FDDC. This increased chloride load in DCT fails to get reabsorbed, due to the basic pathophysiology of GS, resulting in a marked reduction of DFCR. On the other hand thiazide only mildly increases the FDDC and mildly depresses the DFCR. Therefore, use of the furosemide loading test localizes the defect in GS to a distal tubular location and the thiazide loading test nails the location to the thiazide-sensitive NCC. This observation is of immense importance in a resource poor setting like India, where this study was located, and where genetic testing for a disease like GS is practically out of the question for most patients. These two simple and easy to perform bedside tests can easily clinch the diagnosis of GS. Differentiation of GS from BS using one or the other tests has been reviewed earlier.[3]
We also propose the use of the normogram [Figure 1], which originally appeared in the classic article by Gill and Bartter.[10] The placement of basal values close to the surreptious vomiting (SV) group, a marked change effected by the furosemide loading test (FLT) group, and the minimal change caused by the thiazide loading test (TLT) group may be used as a simple graphical measure to diagnose Gitelman's syndrome. The only two other case reports from India[1213] did not make use of the renal clearance studies used in this article. To the best of our knowledge, this is probably the first reported case of Gitelman's syndrome in India, where these two simple techniques have been used to localize the site of the renal tubular defect and to confirm the diagnosis.[14]
Source of Support: Nil
Conflict of Interest: None declared.
References
- Possible discriminations of Gitelman's syndrome from Bartter's syndrome by renal clearance study: Report of two cases. Am J Kidney Dis. 1995;25:637-41.
- [Google Scholar]
- Gitelman's variant of Bartter's syndrome, inherited hypokalemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Clcotransporter. Nature Genet. 1996;12:24-30.
- [Google Scholar]
- Abnormal reabsorption of Na+/ Cl_ by the thiazide-inhibitable transporter of the distal convoluted tubule in Gitelman's syndrome. Am J Nephrol. 1997;17:103-11.
- [Google Scholar]
- From bench to bedside: Diagnosis of Gitelman's syndrome – defect of sodium-chloride cotransporter in renal tissue. Kidney Int. 2006;70:813-7.
- [Google Scholar]
- Fractional excretion of potassium in normal subjects and in patients with hypokalemia. Postgrad Med J. 1995;71:211-2.
- [Google Scholar]
- A new familial disorder characterized by hypokalemia and hypomagnesaemia. Trans Assoc Am Physicians. 1966;79:221-3.
- [Google Scholar]
- Novel molecular variants of the Na–Clcotransporter gene are responsible for Gitelman syndrome. Am J Hum Genet. 1996;59:1019-26.
- [Google Scholar]
- Evidence for a prostaglandin-independent defect in chloride reabsorption in the loop of Henle as a proximal cause of Bartter syndrome. Am J Med. 1978;65:766.
- [Google Scholar]
- Attenuated renal excretion in response to thiazide diuretics in Gitelman's syndrome: A case report. J Korean Med Sci. 2002;17:567-70.
- [Google Scholar]
- Gitelman's syndrome presenting as recurrent paralytic ileus due to chronic renal tubular K+ wasting. J Assoc Physicians India. 2010;58:322-4.
- [Google Scholar]




