

Translate this page into:
Fibrillary Glomerulonephritis with Prevalent IgA Deposition Associated with Psoriasis
-
Received: ,
Accepted: ,
How to cite this article: Patel S, Patel H, Kunpara J, Bhalodiya R, Shah J, Patwari D, et al. Fibrillary Glomerulonephritis with Prevalent IgA Deposition Associated with Psoriasis. Indian J Nephrol. doi: 10.4103/ijn.ijn_93_23
Abstract
Fibrillary and immunotactoid glomerulonephritis represent the pathological entities characterized by structured fibrillary/microtubular deposits, whose identification is possible only by electron microscopy. We report a 46-year-old female who presented with proteinuria 15 years after the onset of psoriasis. Diffuse global glomerulosclerosis pattern was noted on light microscopy. In immunofluorescence microscopy, predominant IgA deposition was observed and electron microscopy showed organized randomly arranged fibrillary deposits with diameter ranging between 10 and 23 nm in mesangial, subendothelial, intramembranous, and subepithelial sites.
Keywords
Fibrillary glomerulonephritis
IgA deposition
psoriasis

Introduction
In 1977, Rosenmann and Eliakim reported a glomerulopathy characterized by infiltration of glomeruli by a fibrillar material that resembled amyloid, but did not stain with Congo red.1 Following this precedent, Alpers et al.2 adopted the term fibrillary glomerulonephritis. The most common forms of nonamyloid glomerular deposition diseases are fibrillary glomerulonephritis (FGN) and immunotactoid glomerulopathy (ITGN).3 Proteomic studies have identified DnaJ heat shock protein family (Hsp40) member B9 (DNAJB9) as a potential novel, highly specific biomarker for the diagnosis of FGN.4 FGN has been associated with malignancy, monoclonal gammopathy, autoimmune disease, or infection in 30%– 50% of cases.5 Rare cases of IgA nephropathy (IgAN) associated with Congo red–negative FGN have been reported.6-8
We describe a case of psoriasis who presented with nephrotic range proteinuria and microscopic hematuria 15 years after the apparent onset of the disease. Renal involvement was peculiar and showed diffuse global glomerulosclerosis with IgA deposits with fibrillary glomerular deposition.
Case Report
A 46-year-old woman presented with complaints of ankle pain for the last 6 months and lower limb edema for 2 months. The patient has been a known case of psoriasis for the last 15 years; she was off treatment for the last 2 years and had hypertension for the last 5 years. She had a history of treatment with local steroid ointment for skin lesions. She had no complaints of joint pain, fever, skin rashes, oral ulcer, or photosensitivity.
On examination, she was afebrile and her pulse rate was 84/min and respiratory rate was 16/min. She had high blood pressure (BP; 190/100 mmHg), pedal edema, and hypopigmented patches. There was no jaundice or lymphadenopathy. Systemic examination was normal.
Investigations showed: hemoglobin (Hb): 9.8 g/dl, total leukocyte count (TLC): 8400/µl, platelet count: 2.83 lakhs, sodium: 139 mEq/l, potassium: 5.0 mEq/l, creatinine: 2.64 mg/dl, total calcium: 8.1 mg/dl, and uric acid: 5.9 mg/dl. Urine examination showed protein: 3+, red blood cells (RBCs): 8–10/hpf (70% dysmorphic), pus cell: 2–3/hpf, and urine protein creatinine ratio was 12.1 mg/mg. Complement (C3) level was 149 mg/dl, cholesterol was 279 mg/dl, and Antinuclear antibody (ANA) by Immunofluorescence (IF) was negative on 1:160 dilution. Hepatitis B (Hep B), hepatitis C (Hep C), and human immunodeficiency virus (HIV) were negative. Ultrasonography showed normal-sized kidneys.
Light microscopic examination of kidney biopsy showed that all nine glomeruli were completely sclerosed with few foci of tubular atrophy and mild interstitial mononuclear inflammatory cell infiltrate [Figure 1a]. Congo red was negative. On immunofluorescence microscopy, all glomeruli showed an intense deposition of IgA (3+) with IgG, kappa, and lambda localized in the mesangium as well as in the capillary [Figure 1b and c], whereas IgM, C3, C1q, and fibrinogen were negative.
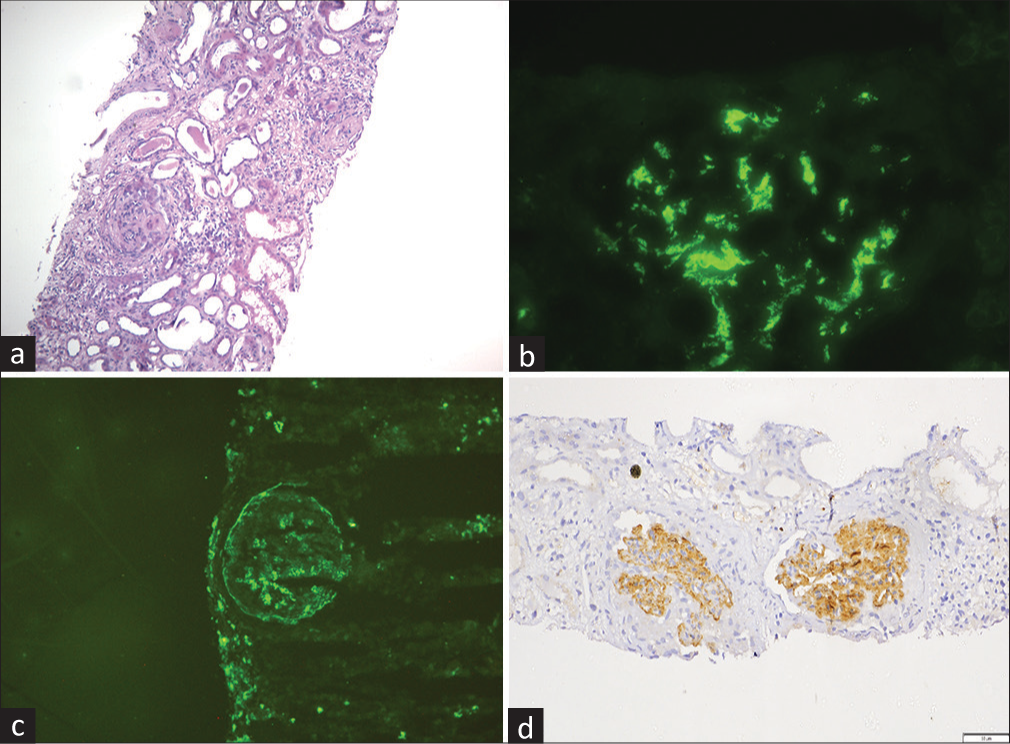
- (a) Two completely sclerosed glomeruli with mild interstitial mononuclear inflammatory cell infiltrate (light microscopy, H and E stain, 10×). (b) Intense IgA deposition in the mesangium and in the capillary walls (immunofluorescence, IgA antiserum, 40×). (c) IgG deposition in the mesangium and in the capillary walls (immunofluorescence, IgG antiserum, 40×). (d) Immunohistochemistry shows bright staining for DNAJB9 in the mesangium and the capillary walls (immunohistochemistry, 20×), DNAJB9 = DnaJ heat shock protein family (Hsp40) member B9, H and E = hematoxylin and eosin.
On electron microscopy, three glomeruli showed marked thickening and duplication of basement membrane and diffuse effacement of foot process of podocytes with condensation of actin-based cytoskeleton. There were numerous granular electron-dense intramembranous and mesangial deposits with focal subendothelial deposits. These deposits showed fibrillary substructure on high power with cross diameter ranging from 17 to 22 mm, which was DNAJB9 positive on Immunohistochemistry (IHC) [Figures 1d and 2a, b]. The final diagnosis was made as FGN. After diagnosis, she was treated with steroid and mycophenolate mofetil. Her renal function remains stable at 8 months of follow-up.
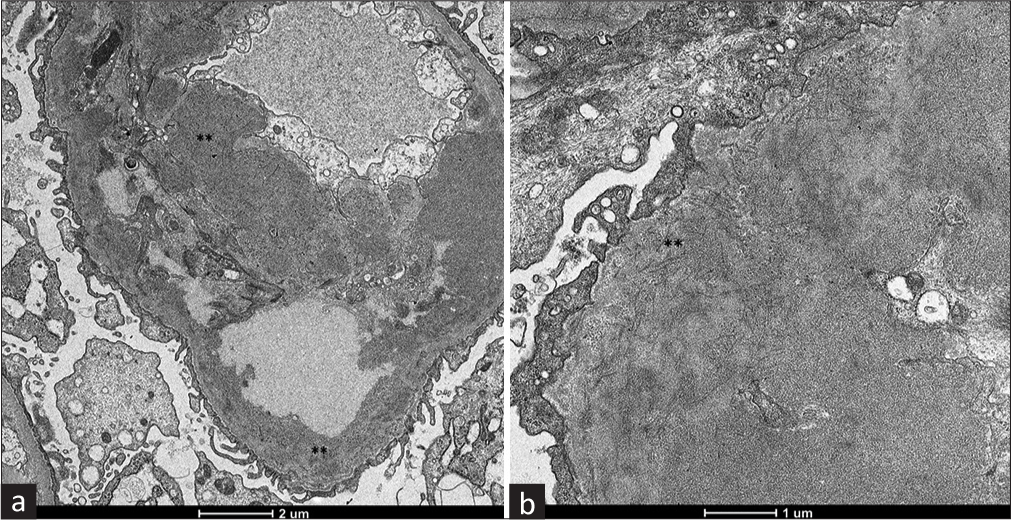
- (a) Electron-dense deposits located in the basement membrane and mesangium (**). (electron microscopy, 2600×). (b) Mesangial deposits composed primarily of short and rigid fibrils without a hollow core, with a diameter ranging between 10 and 23 nm (**), contained in dense fine granular material (electron microscopy, 8500×).
Discussion
This case report describes a rare association of psoriasis with IgA-predominant FGN. Most cases of FGN were previously considered as idiopathic. However, a case series of 66 FGN patients by Nasr et al.5 showed that 23% of patients had malignancy (most commonly, carcinoma), 17% had dysproteinemia, and 15% had autoimmune disease (most commonly, Crohn’s disease, systemic lupus erythematosus [SLE], Graves’ disease, and idiopathic thrombocytopenic purpura). Cases of FGN associated with Sjögren’s syndrome,9 rheumatoid arthritis,10 and Behçet syndrome11 had also been described. Therefore, unlike the above-mentioned associations, the association of FGN and underlying psoriasis represents a novel association.
There is usually predominant deposition of some IgG subclasses like IgG4 and, to a lesser extent, IgG1 in IHC.4 The association of FGN with IgA deposition, as observed in our case, has been previously published, but it is a rare event.6-8 In a case series by Nasr et al.,5 all 66 cases showed glomerular positivity for IgG, with a mean intensity of 2.5+ and 28% and 47% of cases were positive for IgA and IgM, respectively, with a mean intensity of 1.0+. The glomerular staining for IgM and IgA was weaker than IgG in all cases except one, in which IgA staining was stronger than IgG.
In our case, IHC was positive for DNAJB9. Co-localization of staining for DNAJB9 and IgG within glomerular deposits suggests that DNAJB9 could represent a putative autoantigen in FGN. However, a circulating DNAJB9 autoantibody has not been identified in these patients.4 Another possibility is that DNAJB9 may be a protein that secondarily binds to misfolded IgG molecules by recognizing aggregation-prone motifs. This hypothesis could explain the poor response to rituximab and other immunosuppressive agents in patients with FGN.
The presenting clinical features of FGN are similar to other forms of glomerular diseases. In a case series of 186 patients with FGN, the following findings were noted on presentation:12 hematuria in 70%, proteinuria in 100% with nephrotic syndrome (protein excretion ≥3.5 g/day) in 70%–75%, kidney function impairment (serum creatinine ≥1.5 mg/dl) in 50%–55%, and hypertension in 65%–70%, which is the same as our case who presented with nephrotic range proteinuria.
In the present case, though IF was suggestive of IgA nephropathy, we did Electron microscopy (EM), which changed the final diagnosis to FGN, showing the importance of EM. FGN itself is different from other forms of glomerulonephritis in terms of prognosis, treatment, and posttransplant recurrence.
Patients with FGN are difficult to treat, and there are no randomized controlled trials to guide optimal therapy. Cases where secondary causes are identified, such as malignancy, monoclonal gammopathy, infection, or autoimmune disease, may benefit from treatment of the underlying disorder.
Patients who do not have a secondary cause of FGN and have an estimated glomerular filtration rate (GFR) ≥60 ml/min/1.73 m2 and proteinuria <3.5 g/day can be managed conservatively with antiproteinuric therapy like angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs), BP control, and dietary sodium restriction. In patients with an estimated GFR <60 ml/min/1.73 m2 or proteinuria >3.5 g/day, rituximab can be used. Some but not all reports suggest that rituximab may be associated with complete or partial remission of proteinuria in patients with FGN.5,13,14 Immunosuppressive therapy such as glucocorticoids along with cyclophosphamide, mycophenolate mofetil, cyclosporine, melphalan, azathioprine, and rapamycin has been reported in uncontrolled studies with limited results.3,5
Approximately 40%–50% of patients reported with FGN develop End stage kidney disease (ESKD) within 2–6 years.3,5 Dialysis or kidney transplantation can be performed in patients with FGN who progress to ESKD. With kidney transplantation, recurrent disease can develop in the allograft,5,12,15 but the rate of progression is usually slower than in the native kidney. The rate of recurrence appears to be higher in patients who have a monoclonal gammopathy.15 A case series by El Ters et al.16 evaluated 14 patients with DNAJB9-positive FGN who underwent kidney transplantation and had protocol allograft biopsies at 4, 12, 24, 60, and 120 months posttransplantation. The median follow-up was 5.7 years for 14 patients, with three (21%) patients showing recurrence of FGN detected on the 5-year (n = 1) and 10-year (n = 2) allograft biopsies. Median time to recurrence was 10.2 (interquartile range [IQR]: 5–10.5) years. The remaining 11 patients had no evidence of histologic recurrence on the last posttransplantation biopsy, although the median time of follow-up was significantly less at 4.4 years.
Conclusion
This case showed a rare combination of FGN and prevalent IgA deposition, in the clinical context of psoriasis. It also emphasizes the significance of the electron microscopy in the identification of these uncommon entities.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
Nil.
References
- Nephrotic syndrome associated with amyloid-like glomerular deposits. Nephron. 1977;18:301-8.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrillary glomerulonephritis: An entity with unusual immunofluorescence features. Kidney Int. 1987;31:781-9.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrillary and immunotactoid glomerulonephritis: Distinct entities with different clinical and pathologic features. Kidney Int. 2003;63:1450-61.
- [CrossRef] [PubMed] [Google Scholar]
- DnaJ heat shock protein family B member 9 is a novel biomarker for fibrillary GN. J Am Soc Nephrol. 2018;29:51-6.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrillary glomerulonephritis: A report of 66 cases from a single institution. Clin J Am Soc Nephrol. 2011;6:775-84.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrillary-immunotactoid glomerulopathy with renal deposits of IgAlambda: A rare cause of glomerulonephritis. Clin Nephrol. 1999;52:383-9.
- [Google Scholar]
- Immunoelectron microscopic analysis of intraglomerular deposits in IgA-dominant immunotactoid glomerulopathy. The Japanese journal of Clinical Pathology 2002. ;. ;50:1085-9.
- [Google Scholar]
- A case of immunotactoid glomerulopathy with IgA2, kappa deposition ameliorated by steroid therapy. Nihon Jinzo Gakkai Shi. 2003;45:449-56.
- [Google Scholar]
- Fibrillary glomerulonephritis in a patient with Sjogren's syndrome. Cureus. 2018;10:e2483. doi: 10.7759/cureus.2483
- [CrossRef] [Google Scholar]
- Fibrillary glomerulonephritis in rheumatoid arthritis. Nephrology. 2010;15:266-7.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrillary glomerulonephritis associated with Behçet's syndrome. Ren Fail. 2012;34:637-9.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical features, predictors of disease progression and results of renal transplantation in fibrillary/immunotactoid glomerulopathy. Nephrol Dial Transplant. 1996;11:837-42.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab treatment of fibrillary glomerulonephritis. Am J Kidney Dis. 2008;52:1158-62.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab treatment for fibrillary glomerulonephritis. Nephrol Dial Transplant. 2014;29:1925-31.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term outcome of kidney transplantation in patients with fibrillary glomerulonephritis or monoclonal gammopathy with fibrillary deposits. Kidney Int. 2009;75:420-7.
- [CrossRef] [PubMed] [Google Scholar]
- Recurrence of DNAJB9-positive fibrillary glomerulonephritis after kidney transplantation: A case series. Am J Kidney Dis. 2020;76:500-10.
- [CrossRef] [PubMed] [Google Scholar]




