

Translate this page into:
GALNT3 Mutation in Hyperphosphatemic Familial Tumoral Calcinosis – Novel Etiology of Secondary Amyloidosis
Corresponding author: Sourabh Sharma, Department of Nephrology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India. E-mail: drsourabh05@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Sharma S, Arora S, Binoy R, Agarwal A, Prasad P, Verma H. GALNT3 Mutation in Hyperphosphatemic Familial Tumoral Calcinosis – Novel Etiology of Secondary Amyloidosis. Indian J Nephrol. 2025;35:303-5. doi: 10.25259/ijn_534_23
Abstract
Tumoral calcinosis is a rare syndrome characterized by calcium salt deposition in different periarticular soft tissue regions. We report this case of tumoral calcinosis with history of persistent soft tissue calcifications for over three decades. He presented with nephrotic syndrome and kidney biopsy revealed secondary amyloidosis. Genetic evaluation revealed GALNT3 mutation and diagnosis of hyperphosphatemic familial tumoral calcinosis was made. With this case report, we want to reiterate the need to consider tumoral calcinosis in secondary amyloidosis differentials and the pivotal role of genetic workup in chronic soft tissue calcifications.
Keywords
Amyloidosis
Hyperphosphatemia
Mutation
Secondary amyloid
Soft tissue calcification
Tumoral calcinosis

Introduction
Tumoral calcinosis (TC), a rare clinical syndrome characterized by periarticular soft tissue calcifications, presents a diagnostic challenge due to its varied manifestations.1,2 We present a case of amyloidosis secondary to long-standing soft tissue calcification, eventually diagnosed to be hyperphosphatemic familial TC. To the best of our knowledge, this is the first reported biopsy-proven case of secondary amyloidosis presenting as nephrotic syndrome attributable to primary TC.
Case Report
A 39-year-old male, born out of a consanguineous marriage, first noticed a lump at his left hip at the age of 10 years. An excision biopsy revealed fibro-collagenous tissue with multiple calcific deposits with lymphoplasmacytic infiltrates, and he was diagnosed to have TC based on radiological and histopathological findings. No family history of similar illness was present. He was found to have hyperphosphatemia with normal serum calcium levels. On further evaluation, his 25-hydroxy vitamin D was found to be 44 nmol/L, intact parathormone (iPTH) 5 pg/mL, and FGF-23 13 RU/mL (reference: 0–150). His blood sugar and renal function were within normal range. He was started on acetazolamide, but his serum phosphorus levels remained high (ranging 5–7 mg/dL). For the next 30 years, he continued to develop soft-tissue calcification in different body areas. He subsequently underwent corrective surgeries and remained on a low-phosphate diet, acetazolamide, and aluminum hydroxide.
He was presented to our department with pedal and facial swelling for 1 week, along with frothuria. There was no history of nocturia, hematuria, lithuria, skin rash, fever, joint pains, alopecia, or impotency. The patient was normotensive, and on laboratory evaluation, he was found to have nephrotic syndrome. The serum creatinine was 1.5 mg/dL, urinalysis revealed 4 + albumin without active sediment, 24-h proteinuria was 5 g/day, and serum total protein and albumin were 5 g/dL and 1.5 g/dL, respectively. He had dyslipidemia, with total cholesterol 271 mg/dL. His serum C3 and C4 were normal, Antinuclear antibody (ANA) and anti neutrophil cytoplasmic antibody (ANCA) studies were negative, and viral markers (anti-HIV/HBsAg/anti-HCV) were non-reactive. On repeat metabolic workup, serum calcium was 9.6 mg/dL, serum phosphate was 7.8 mg/dL, iPTH was 16.6 pg/mL, FGF-23 was 7464 RU/mL, 25-hydroxy vitamin D was 28.8 nmol/L, and (1,25)-dihydroxy vitamin D was 12 pmol/L. Furthermore, 24-h urine phosphate was 520 mg/day. Serum protein electrophoresis and serum-free light chain assays were negative. Radiological evaluation (X-ray pelvis and MRI pelvis with contrast) is depicted in Figure 1.
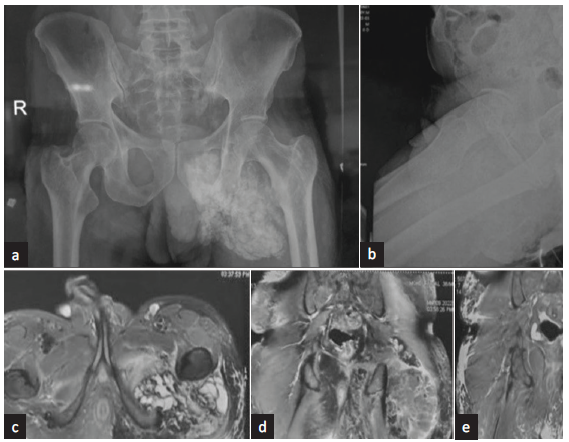
- (a) X-ray pelvis anteroposterior and (b) lateral view depicting heterogenous fluffy opacity involving soft tissues of left side of inferior aspect of left hemi-pelvis and medial aspect of upper thigh, overlying ischial bones, extending up to left side hemi-scrotum. Magnetic resonance imaging (MRI) Pelvis with (c) contrast axial and (d) and (e) coronal view depicts large multilobulated septate cystic, partly solid soft tissue mass in proximal thigh & adjacent buttock surrounding posterior aspect of intertrochanteric and proximal shaft of femur. Septate cystic area was hypointense in T1, and hypo to hyperintense in T2 with multiple air fluid level showing dependent calcium. There was post-contrast patchy peripheral enhancement of the mass.
A kidney biopsy [Figure 2] showed glomerular deposition of amorphous extracellular material, which was PAS and silver negative and showed congophilia. Serum amyloid A staining was positive on immunohistochemistry. A diagnosis of secondary amyloidosis was made, and investigations were done to rule out secondary causes. Rheumatoid factor was negative (Mantoux test), and contrast enhanced computerised tomography (CECT) chest and abdomen showed no obvious abnormality. With no other cause being found, amyloidosis was attributed to TC. On genetic evaluation, the patient was found to have GALNT3 mutation (homozygous; variant c.1459T>C; location: Exon 8 and autosomal recessive inheritance). He was finally labeled as having hyperphosphatemic familial tumoral calcinosis (TC). On follow-up, his serum phosphate level did not decline on the maximum dose of sevelamer and was shifted to lanthanum carbonate. He underwent a lumpectomy after stabilization. Presently, he is normophosphatemic on phosphate binder (lanthanum carbonate) and stable.
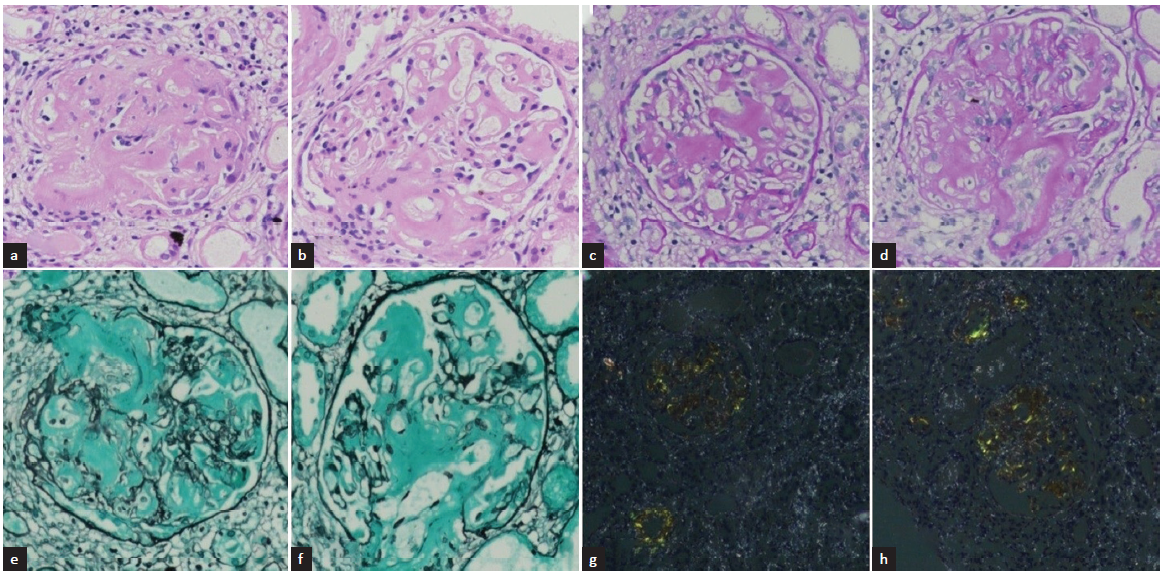
- Kidney biopsy - light microscopy in 20X magnification- (a and b) Hematoxylin and Eosin staining show glomerulus with moderate mesangial matrix expansion due to deposition of amorphous extracellular material. (c and d) Periodic acid-schiff (PAS) staining depicts that amorphous extracellular material is PAS positive. (e and f) Silver methenamine staining depicts that the material is silver negative. (g and h) Congo red staining show congophilic deposits with apple-green birefringence under polarized microscope.
Discussion
TC, also known as Teutschlander disease,1 is a rare syndrome characterized by calcium salt deposition in different periarticular soft-tissue regions.2 The disorder was first stated by Inclan et al. in 1943.3 It manifests in childhood or adolescence as painless, firm, tumor-like masses around the joints. It is divided into two varieties: primary and secondary.1,2,4 The primary variety has two subtypes: hyperphosphatemic type with familial basis: mutations in GALNT3, KLOTHO, or FGF23 genes; and normophosphatemic type with mutation in the SAMD9 gene.
The secondary variety is mainly associated with chronic kidney disease. Diagnosis is mainly based on imaging modalities. Primary treatment is surgical, but there is high recurrence rate. The hyperphosphatemic variety warrants medical management (phosphate binders/acetazolamide) prior to surgery. The next step for non-responders is subtotal or total parathyroidectomy.1,2,4
Secondary amyloidosis is a long-term complication of several chronic inflammatory disorders. Organ damage results from the extracellular deposition of proteolytic fragments of serum amyloid A.5 There has been only a single case report of TC leading to amyloidosis.6 A kidney biopsy was not performed in this case, and amyloidosis was diagnosed by rectal biopsy. Our case is the first reported biopsy-proven case of secondary amyloidosis presenting as nephrotic syndrome attributable to primary TC (hyperphosphatemic; GALNT3 mutation).
This case report imparts three key learning points. First, it underscores the importance of considering TC in differentials of secondary amyloidosis. Second, it highlights a potential long-term complication of unaddressed TC. Lastly, it emphasizes the pivotal role of genetic analysis in cases of chronic soft-tissue calcifications.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
References
- A rare benign differential diagnosis of proliferative space-occupying lesions in the periarticular soft tissue. Orthopade. 2004;33:829-35.
- [CrossRef] [PubMed] [Google Scholar]
- Review of tumoral calcinosis: A rare clinico-pathological entity. World J Clin Cases. 2014;2:409-14.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Secondary, AA, amyloidosis. Rheum Dis Clin North Am. 2018;44:585-603.
- [CrossRef] [PubMed] [Google Scholar]
- Systemic amyloidosis secondary to tumoral calcinosis. Nephron. 1991;59:525-6.
- [CrossRef] [PubMed] [Google Scholar]




