

Translate this page into:
Hidden in Plain Sight: An Unusual Cause of Rapidly Progressive Renal Failure
Address for correspondence: Dr. S. V. Vyahalkar, Department of Nephrology, Grant Medical College and Sir JJ Group of Hospitals, Mumbai - 400 008, Maharashtra, India. E-mail: drsameer2010@gmail.com
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Hyperoxaluria and resultant oxalate nephropathy are infrequently reported causes of irreversible renal failure. A rapid decline in renal function in an otherwise insidiously progressive oxalate nephropathy may be triggered by various superimposed insults like the use of nephrotoxic drugs. We present the case of a patient with rapidly progressive renal failure due to oxalate nephropathy that lead to a retrospective diagnosis of chronic pancreatitis. This case highlights the importance of timely assessment for enteric hyperoxaluria in patients with unexplained renal failure of tubulointerstitial nature.
Keywords
Chronic pancreatitis
enteric hyperoxaluria
oxalate nephropathy
renal failure

Introduction
Increased intestinal absorption or increased endogenous synthesis of oxalate leads to hyperoxaluria. Apart from the well-known complications of nephrocalcinosis and nephrolithiasis, hyperoxaluria can also cause tubulointerstitial nephritis (TIN) that has high risk of progression to chronic kidney disease (CKD).
Case Report
A 32-year-old male was admitted for renal failure. Five years ago, diabetes mellitus (DM) had been diagnosed on the evaluation of unintentional weight loss. Six months before present admission, pulmonary tuberculosis (PTB) was diagnosed. Investigations at that time including kidney and liver function tests, complete blood count, and urine analysis were normal; X-ray chest revealed bilateral reticulonodular infiltrates and small thin-walled cavities; sputum smear for acid-fast bacilli was negative; computed tomography (CT) scan of thorax revealed bilateral multiple centrilobular nodules with adjacent ground glass opacities, fibro-bronchiectatic changes in upper lobes and multiple necrotic mediastinal lymph nodes. Since there was past history of PTB with drug default, antitubercular treatment (ATT) was started as per category II of DOTS (thrice a week combined regime of streptomycin for 2 months, pyrazinamide for 3 months, and isoniazid, rifampicin, and ethambutol for 8 months). Three months ago, progressive increase in serum creatinine was noted, and he was investigated at another hospital. Serum creatinine was 7.1 mg/dl, complete blood count, liver function tests, and urine analysis were normal; 24 h urine protein was 100 mg; tests for HIV, hepatitis B and C virus infection were negative, and ultrasonography of kidney showed normal-sized kidneys. Kidney biopsy was performed; light microscopy had revealed only one glomerulus with normal morphology, mild interstitial edema with lymphocytic and neutrophilic infiltrates and few tubules with changes of acute injury and oxalate crystal deposition. Dose-modified rifampicin-free ATT had been continued, however, renal function had declined further. One month before the present admission, hemodialysis was initiated due to uremic symptoms. Table 1 summarizes the trend of renal function. In the present admission, investigations were as follows: Hb 9.5 g/dl, WBCs 7800/cmm, platelets 2.4 lac/cmm, serum. creatinine 11.4 mg/dl, total bilirubin 0.6 mg/dl, SGOT 18 IU/L, SGPT 38 IU/L, serum calcium 8.8 mg/dl, serum phosphorus 5.8 mg/dl; Urine analysis-trace protein, 1–2 red blood cells, and 3–4 pus cells/hpf, no crystalluria; 24 h urine protein-94 mg. Fundoscopy revealed changes of mild non-proliferative diabetic retinopathy. Serum complement level was normal, antinuclear antibody and anti-neutrophil cytoplasmic antibody tests were negative. Kidney biopsy was repeated; it revealed 14 viable glomeruli with mild mesangial expansion without evidence of hypercellularity, nodular glomerulosclerosis, or glomerular basement membrane (GBM) alterations [Figure 1a]. Tubules showed extensive inspissation of refractile calcium oxalate (CaOx) crystals in tubular epithelial cells and lumina with acute tubular injury [Figure 1b]. The crystals showed strong birefringence under polarized light. Mild interstitial edema was observed with lymphocytic infiltrates; tubular atrophy and interstitial fibrosis in 25% of the cortex. Eosinophilic infiltrates or acute tubular necrosis was not seen. Immunofluorescence was negative. Electron microscopy showed mild thickening of GBM (mean thickness 475.3 nm). Electron-dense deposits were absent. Diagnosis of oxalate nephropathy with mild diabetic nephropathy (Class IIa) was made. Urine Oxalate:Creatinine ratio was 41 mg/g (normal <32) indicating hyperoxaluria. He had no clinical features of primary hyperoxaluria. He had no history of renal calculi, recurrent abdominal pain, diarrhea, abdominal surgery, recurrent respiratory tract infections or infertility, consumption of herbal or nonprescription medicines, or alcohol consumption. Due to the presence of DM and significant weight loss, chronic pancreatitis (CP) was suspected.[1] Further investigations revealed: Serum amylase 96 IU/L (normal 25–140), lipase (325 IU/L, normal 40–190), CT abdomen revealed diffuse calcification of head [Figure 2a], body and tail of pancreas. Magnetic resonance cholangiopancreatography confirmed severe atrophy of pancreas with multiple calcific foci in the parenchyma and pancreatic duct [Figure 2b]. Fecal elastase-1 level was low (90 μg/g of stool, normal >200). These features were diagnostic of chronic calcific pancreatitis[1] and hyperoxaluria. He was treated with pancreatic enzyme supplementation, calcium carbonate, sodium bicarbonate, pyridoxine, and increased fluid intake for oxalate nephropathy. ATT was continued without rifampicin. However, the patient remained dialysis dependent at 6-month follow-up.

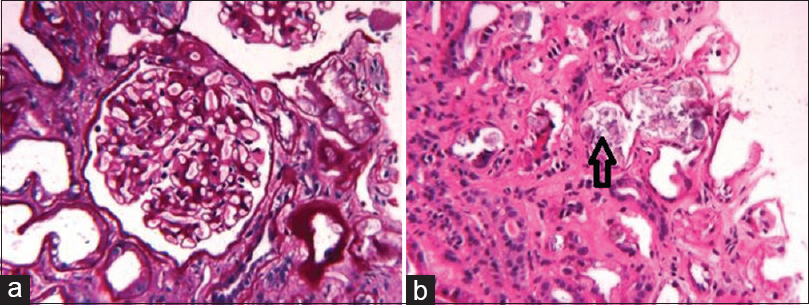
- (a) Periodic acid–Schiff stain reveals mild mesangial expansion without hypercellularity and normal glomerular basement morphology (b) Calcium oxalate crystals in lumen of renal tubule (arrow) on H and E stain
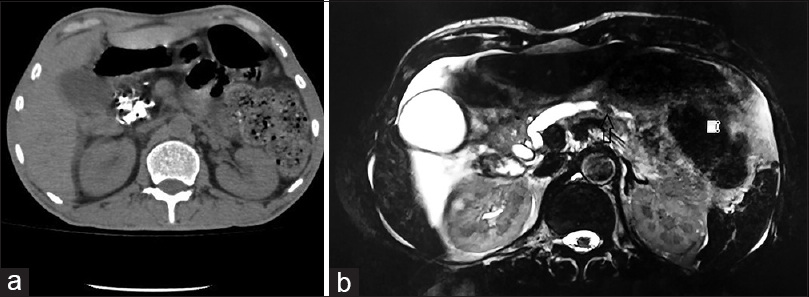
- (a) Computed tomography abdomen shows diffuse calcification of head of pancreas (b) Magnetic resonance cholangiopancreaticogram revealed diffuse pancreatic atrophy with filling defect in the dilated main pancreatic duct suggesting calcification
Discussion
Oxalate is the salt forming ion of oxalic acid that is widely distributed in nature and is excreted primarily by the kidneys through glomerular filtration and tubular secretion. For adults, urinary oxalate excretion over 40–45 mg/day[2] or urine Oxalate:Creatinine ratio more than 32 mg/g[3] is considered as hyperoxaluria. Hyperoxaluria can be classified[245] [Table 2] into primary (due to increased endogenous synthesis of oxalate due to inherited enzyme deficiencies in amino acid metabolic pathway), and secondary (due to increased intestinal absorption from various causes). Enteric hyperoxaluria (EH) refers to a condition in which fat malabsorption causes increased intestinal absorption of oxalate as a result of two main mechanisms, (1) binding of fatty acids with luminal calcium reduces the formation of insoluble CaOx complexes and increases availability of soluble oxalate, and (2) both fatty acids and dihydro bile acids, which occur in high concentration due to short bowel or intestinal bypass surgery, increase the permeability of intestinal mucosa to oxalate. Other mechanisms include reduced intestinal colonization with oxalate degrading bacteria, for example, Oxalobacter formigenes which leads to increased oxalate absorption and deficiency of pyridoxine, a cofactor of the liver enzyme AGXT, that results in increased endogenous synthesis of oxalate.[5] Hyperoxaluria is well-known to cause nephrolithiasis and nephrocalcinosis due to urinary super-saturation of CaOx salt, which is insoluble. In addition, oxalate induced renal injury can also lead to TIN, interstitial fibrosis and CKD.[5] The NOD-like receptor family, pyrin domain containing-3 (NLRP-3) inflammasome pathway has been implicated in the pathogenesis of progressive renal injury due to CaOx. The NLRs are a set of intracellular pattern recognition receptors, which after interaction with CaOx, assemble to form caspase-1 activating platforms called inflammasomes that control the maturation and secretion of interleukin (IL)-1 β and IL-18, resulting in inflammatory cascade.[67] Other putative mechanisms of CaOx-induced renal injury include direct cytotoxicity through activation of the necroptosis signaling pathway and activation of reactive oxygen species.[8]

Acute oxalate nephropathy (AON) is characterised by rapidly progressive renal failure and evidence of TIN with abundant deposits of oxalate within tubular lumina, tubular epithelial cells and/or interstitial space on kidney biopsy.[9] Reports of AON in association with CP are rare, most of the patients have a preexisting diagnosis of CP and the prognosis is dismal with almost half of the reported patients reaching end-stage renal disease.[910] The present report is unusual also due to the fact that the patient had asymptomatic CP. In a recent observational study of prevalent patients of CP, half of which were already on pancreatic enzyme supplementation, hyperoxaluria was found in 23% and factors such as steatorrhea, pancreatic atrophy on imaging, and fecal elastase-1 level <100 μg/g of stool were strongly associated with hyperoxaluria and progressive decline in GFR.[11] Increase in steatorrhea, hypovolemia, metabolic acidosis, nephrotoxic drugs, discontinuation of pancreatic enzyme therapy in patients with CP and progression of underlying CKD can lead to acute deterioration of renal function in EH.[910] AON due to EH mostly occurs in patients with preexisting CKD, mainly related to diabetic nephropathy and has worse prognosis in this population.[1213] Our patient also had histological evidence of diabetic nephropathy along with oxalate nephropathy. We also considered the possibility that nephrotoxicity of anti-tubercular drugs might have contributed to the worsening of renal function in our patient. However, we believe that oxalate nephropathy was primarily responsible because (1) histological features of oxalate nephropathy were present, (2) features associated with drug-induced interstitial disease such as tubulitis, tubular necrosis, and extensive interstitial inflammatory cell infiltrates were not seen, (3) discontinuation of rifampicin therapy did not improve renal function,[14] and (4) use of Streptomycin, the least nephrotoxic among aminoglycosides, is a distinctly uncommon cause of irreversible renal failure.[15]
Conclusion
Histological evidence of oxalate crystal deposition warrants search for etiological diagnosis of hyperoxaluria. Evaluation for oxaluria and malabsorption should be considered early for diagnosis of EH in patients with acute kidney injury and CKD of tubulointerstitial nature. Conversely, close monitoring of renal function and oxaluria should be performed in patients with CP.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- A proposal for a new clinical classification of chronic pancreatitis. BMC Gastroenterol. 2009;9:93.
- [Google Scholar]
- Histopathological occurrence and characterisation of calcium oxalate: A review. J Clin Pathol. 1977;30:800-11.
- [Google Scholar]
- Enteric hyperoxaluria: An important cause of end-stage kidney disease. Nephrol Dial Transplant. 2016;31:375-82.
- [Google Scholar]
- Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest. 2013;123:236-46.
- [Google Scholar]
- NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int. 2013;84:895-901.
- [Google Scholar]
- Crystal nephropathies: Mechanisms of crystal-induced kidney injury. Nat Rev Nephrol. 2017;13:226-40.
- [Google Scholar]
- Oxalate nephropathy associated with chronic pancreatitis. Clin J Am Soc Nephrol. 2011;6:1895-902.
- [Google Scholar]
- Acute deterioration of renal function associated with enteric hyperoxaluria. Clin Nephrol. 1990;34:116-21.
- [Google Scholar]
- Enteric hyperoxaluria in chronic pancreatitis. Medicine (Baltimore). 2017;96:e6758.
- [Google Scholar]
- Oxalate nephropathy complicating Roux-en-Y Gastric Bypass: An underrecognized cause of irreversible renal failure. Clin J Am Soc Nephrol. 2008;3:1676-83.
- [Google Scholar]
- Rapidly progressive irreversible renal failure in patients with pancreatic insufficiency. Am J Kidney Dis. 2003;42:842-5.
- [Google Scholar]
- Acute tubulointerstitial nephritis related to antituberculous drug therapy. Clin Nephrol. 2010;73:413-9.
- [Google Scholar]
- Antibiotic and immunosuppression-related renal failure. In: Coffman TM, Falk RJ, Molitoris BA, Neilson EG, Schrier RW, eds. Schrier's Diseases of the Kidney (9th ed). Philadelphia: Lippincott Williams and Wilkins Publishers; 2013. p. :901-2.
- [Google Scholar]




