

Translate this page into:
Hypertension- One ize does not fit all
Address for correspondence: Dr. M. Edwin Fernando, No. 1, Old Jail Road, Old Washermanpet, Chennai - 600 001, Tamil Nadu, India. E-mail: nephroeddy@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Although the vast majority of hypertension is "essential," some may be secondary. And, an accurate diagnosis of secondary cause of hypertension provides the treating clinician with a unique opportunity that renders dramatic response to the patient, either with pharmacologic therapy or surgery. One such secondary cause of hypertension is congenital adrenal hyperplasia due to 11 beta hydroxylase or 17 alpha hydroxylase deficiency. These inherited syndromes are caused by deficient adrenal corticosteroid biosynthesis, in which there is reduced negative feedback inhibition of cortisol and, depending on the steroidogenic pathway involved, an alteration in adrenal mineralocortiocoid and androgen secretion occurs. Here, we present, a young adult presented with hypertension, in association with hypokalemia and metabolic alkalosis, who was diagnosed with congenital adrenal hyperplasia (CAH) due to non-classical variant 11-beta hydroxylase deficiency, which responded dramatically to steroids therapy. Furthermore, we also report two new mis-sense mutations in CYP11B1 gene, a gene coding for 11-betahydroxylase enzyme.
Keywords
11 β hydroxylase deficiency
congenital adrenal hyperplasia
hypertension
hypokalemia

Introduction
Hypertensive variants of congenital adrenal hyperplasia (CAH) occur secondary to 11 β-hydroxylase deficiency and 17 α-hydroxylase deficiency. Both enzyme deficiencies result in reduced cortisol production that results in overproduction of adrenocortiotrophic hormone (ACTH). This raised ACTH stimulates zona fasiculata of adrenal glands to increase the production of steroid precursors with accumulation of 11-deoxycortisone (DOC), a potent mineralocorticoid, leading to hypertension and hypokalemia with suppressed renin and aldosterone levels. In 11 β-hydroxylase deficiency, there is shunting of steroid precursors into androgen pathway, resulting in increased formation of androgens which produce virilisation in females or precocious puberty in males. By contrast, patients with 17 hydroxylase deficiency, with decreased androgen synthesis, present with features of hypogonadism. Here, we describe a patient with hypertension, with associated hypokalemia and metabolic alkalosis.
Case Description
A 22-year old gentleman, with history of treatment for hypertension since 18 years of age, was referred to us for tertiary care evaluation of secondary hypertension.
Ten days prior to presenting to us, he was hospitalized elsewhere for complaints of easy fatigability and generalized weakness following 4–5 episodes of loose stools per day, for 3 days. His medical records revealed that, on admission, his blood pressure was 80/60 mm Hg, pulse was 98 bpm, and systemic examination was unremarkable. Laboratory evaluation showed his blood sugar was 98 mg/dL, urea was 24 mg/dL, serum creatinine was 0.9 mg/dL. His serum electrolytes was remarkable for the presence of hypokalemia (potassium = 1.6 mEq/dL). His serum sodium was 141 mEq/dL, chloride was 99 mg/dL, and bicarbonate was 26 mEq/dL. His serum magnesium was 2.1 mg/dL, calcium was 9.1 mg/dL, phosphorus was 1.8 mg/dL, and uric acid was 4.5 mg/dL. His complete blood count showed haemoglobin of 15.0 gm/dl, total white blood cell count of 10,700 cells per cu.mm, differential count showing 64% polymorphs, 26% lymphocytes, 4% eosinophils, and platelet count of 3.4 lakh cells per cu. mm Arterial blood gas analysis showed metabolic alkalosis with respiratory alkalosis (pH- 7.47; HCO3- -27 mEq/dL; pCO2 -34 mm Hg) [Table 1].
| Laboratory investigations | Reference values | On admission (elsewhere) | At discharge (elsewhere) | At our hospital |
|---|---|---|---|---|
| Blood glucose (mg/dL) | 70-100 | 98 | 81 | 88 |
| Blood urea (mg/dL) | 8-21 | 24 | 19 | 24 |
| Serum creatinine (mg/dL) | 0.8-1.3 | 0.9 | 1.0 | 1.0 |
| Serum electrolytes: | ||||
| Sodium (mEq/dL) | 135-145 | 141 | 147 | 143 |
| Potassium (mEq/dL) | 3.5-5.0 | 1.6 | 3.7 | 3.0 |
| Chloride (mEq/dL) | 98-106 | 99 | 101 | 100 |
| Bicarbonate (mEq/dL) | 23-28 | 26 | 22.9 | 28 |
| Magnesium (mEq/dL) | 1.9-2.5 | 2.1 | - | - |
| Calcium (mEq/dL) | 8.8-10.6 | 9.1 | - | - |
| Phosphorus (mg/dL) | 3-4.5 | 1.8 | - | - |
| Uric acid (mg/dL) | 2.8-5.9 | 4.5 | - | - |
| Complete blood count: | ||||
| Haemoglobin (gm/dL) | 12.0-16.0 | 15.0 | 14.6 | 14.67 |
| Total leukocyte count (per µL) | 4500-11,000 | 10,700 | 9600 | 7870 |
| Differential leukocyte count (%) | ||||
| Neutrophils | 40-70 | 64 | 68 | 67 |
| Lymphocytes | 22-44 | 26 | 24 | 28 |
| Eosinophils | 4-11 | 4 | 2 | 5 |
| Monocytes | 0-8 | - | - | - |
| Basophils | 0-3 | - | - | - |
| Platelet count (per µL) | 150,000-400,000 | 304,000 | 30700 | 300,000 |
| Arterial Blood gas analysis: | ||||
| pH | 7.35-7.45 | 7.47 | 7.42 | 7.46 |
| HCO3- (mEq/dL) | 22-26 | 27 | 24 | 28 |
| pCO2 (mm Hg) | 35-45 | 35 | 39 | 34 |
Initial ultrasonography revealed normal sized kidneys bilaterally. He was treated with intravenous 0.9% saline and potassium supplements, following which his dehydration, and serum sodium and potassium were corrected. (147 mEq/dL and 3.7 mEq/dL, respectively). However, during the course of this correction, his hypertension was unmasked, and a blood pressure of 180/90 mm Hg was observed. He was initially started on tablet Nifedipine 30 mg per day. With the blood pressure not under control, his Nifedipine was titrated up to 60 mg per day along with addition of tablet Prazosin 10 mg per day. Tablet Clonidine 0.2 mg per day, was subsequently added to his treatment to control his BP at 160/90 mm Hg. He was also advised to be on salt restricted diet and oral potassium supplements and was referred to us for further evaluation.
On evaluation at our hospital, we noted that he was short statured (height = 153.5 cm) for his age with his weight being 57.3 kg. He did not have pallor nor pedal edema. His blood pressure was 160/90 mm Hg without any significant variation between all four limbs. All his peripheral pulses were felt equally, no special characteristics, normal volume, no radio-radial, no radio-femoral delay. There was no swelling in the region of thyroid, no renal bruit, no evidence of hypertensive retinopathy.
Biochemical investigations at our hospital revealed that his urinalysis was unremarkable. His blood sugar was 88 mg/dL, urea was 24 mg/dL, serum creatinine was 1.0 mg/dL. His serum electrolytes revealed sodium of 143 mEq/dL, potassium of 3.0 mEq/dL, chloride of 100 mg/dL, and bicarbonate of 28 mEq/dL. Arterial blood gas analysis, again, showed metabolic alkalosis with respiratory alkalosis (pH- 7.46; HCO3- -28 mEq/dL; pCO2 -34 mm Hg). His complete blood count showed haemoglobin of 14.6 gm%, total white blood cell count of 7870 cells per cu.mm, differential count showing 67% polymorphs, 28% lymphocytes, 5% eosinophils, and platelet count of 3.0 lakh cells per cu.mm. His thyroid profile was within normal limits [Tables 1 and 2].
| Laboratory investigations | Reference values | |
|---|---|---|
| Plasma renin (microIU/mL) | 4.40-46.10 | 2.94 |
| Serum aldosterone (ng/dL) | 7-30 | 0.97 |
| Serum 17- alpha hydroxyl progesterone (ng/mL) serum | 0.63-2.15 | 4.49 |
| Serum Dehydroepiandrosterone levels (DHEA) (microgm/dL) | 238.4-539.30 | 634.0 |
| Plasma ACTH (pg/mL) | <46.00 | 49.10 |
| At 8 am: | ||
| Serum total cortisol (mcg/dL) | 3-22 | 13.38 |
| Serum 11-deoxy cortisol (ng/dL) | <119 | 8307 |
| Post-ACTH stimulation test: | ||
| Serum total cortisol (mcg/dL) | - | 14.15 |
| Serum 11-deoxy cortisol (ng/dL) | - | 10000 |
Ultrasonography of abdomen showed bilateral normal sized kidneys with normal arterial flow pattern with doppler study. Since hypertension presenting during childhood and adolescence are usually secondary to enzymatic disorders, we were looking for one such cause in our patient. During the current presentation at our hospital, he had hypertension with associated hypokalemia and metabolic alkalosis. Hence, the differentials thought was Liddle syndrome, syndrome of apparent mineralocorticoid excess, glucocorticoid remediable aldosteronism and congenital adrenal hyperplasia due to 11-β Hydroxylase deficiency or 17-α Hydroxylase deficiency. A magnetic resonance imaging of the abdomen was done, which revealed bilateral thickened, wrinkled adrenal glands, with both adrenal limbs measuring more than 4 mm and without any features of neoplasm [Figure 1]. And, his blood investigations highlighted the presence of hyporeninemic hypoaldosteronism, with plasma renin of 2.94 microIU/mL and serum aldosterone of 0.97 ng/dL. In the presence of bilateral thickened adrenal glands, Liddle syndrome was ruled out and absence of high aldosterone levels ruled out, glucocorticoid remediable aldosteronism.
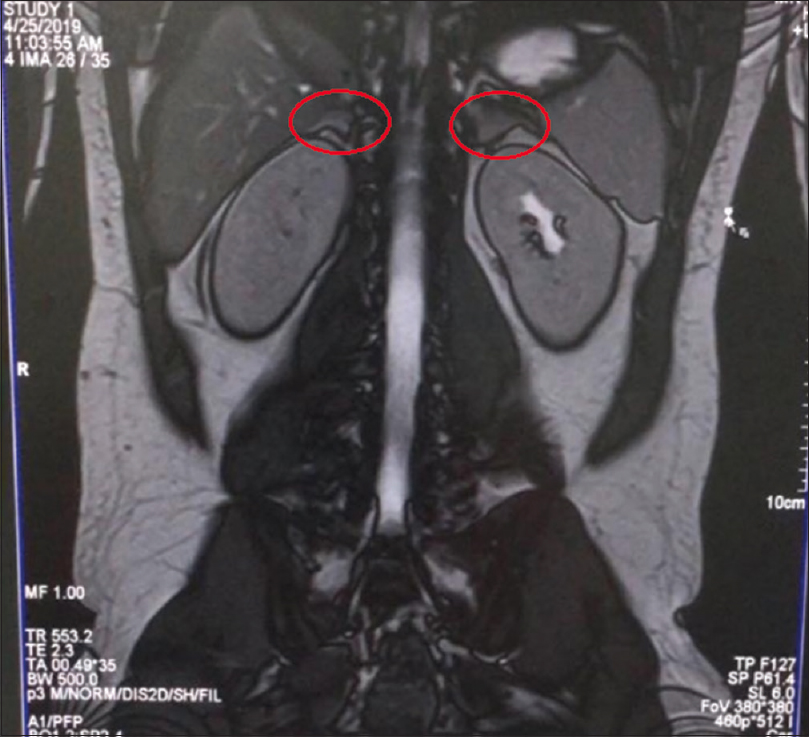
- MRI abdomen showing bilateral thickened and wrinkled adrenal glands
On further workup, he was found to have high serum 17-α hydroxyprogesterone of 4.49 ng/mL, a feature suggestive of CAH. To establish the enzyme deficiency causing CAH, serum dehydroepiandrosterone levels (DHEA) was tested and was found to be high-634.0 microgm/dL, thus making the diagnosis of CAH due to 17-α hydroxylase deficiency unlikely. He had high plasma ACTH - 49.10 pg/mL, which signified the loss of negative feedback control of cortisol resulting in enhanced ACTH-mediated androgen excess. Also, his 8 am serum total cortisol was 13.38 mcg/dL and 11-deoxy cortisol was 8307 ng/dL, which following ACTH stimulation test, revealed values of 14.15 mcg/dL and 10000 ng/dL, thus establishing the diagnosis of CAH due to 11-β Hydroxylase deficiency.
The diagnosis was further corroborated by analysis for gene encoding 11-β Hydroxylase enzyme using targeted gene sequencing. The sequences obtained were aligned to human reference genome (GRCh37/hg19) using Sentieon aligner. It revealed two homozygous missense variants- in exon 3 of the CYP11B1 gene of chromosome 8 that resulted in in the amino acid substitution of Cysteine for Arginine at codon 138 (chr8:g.143958622G>A; Depth: 100×) resulting in (p.Arg138Cys; ENST00000292427.4), and in exon 4 of the CYP11B1 gene on chromosome 8, that resulted in the amino acid substitution of Glutamine for Arginine at codon 208 (chr8:g.143958274C>T; Depth: 82×) (p.Arg208Gln; ENST00000292427.4). [Figure 2a and b]. In view of lack of literature evidence, the variants that were reported as "of uncertain significance" in exon 3 and exon 4 of the CYP11B1 gene were PCR-amplified and the products were sequenced using Sanger sequencing, which validated the existence of the above two mentioned variants that caused the disease condition. He was initiated on T. hydrocortisone 15 mg/day, and other anti-hypertensives and potassium supplementation were slowly withdrawn. He was given vitamin D and calcium supplements in view of long-term requirement of steroids. His blood pressure is currently under control with BP 130/80 mm Hg and is on an OPD-basis follow-ups.
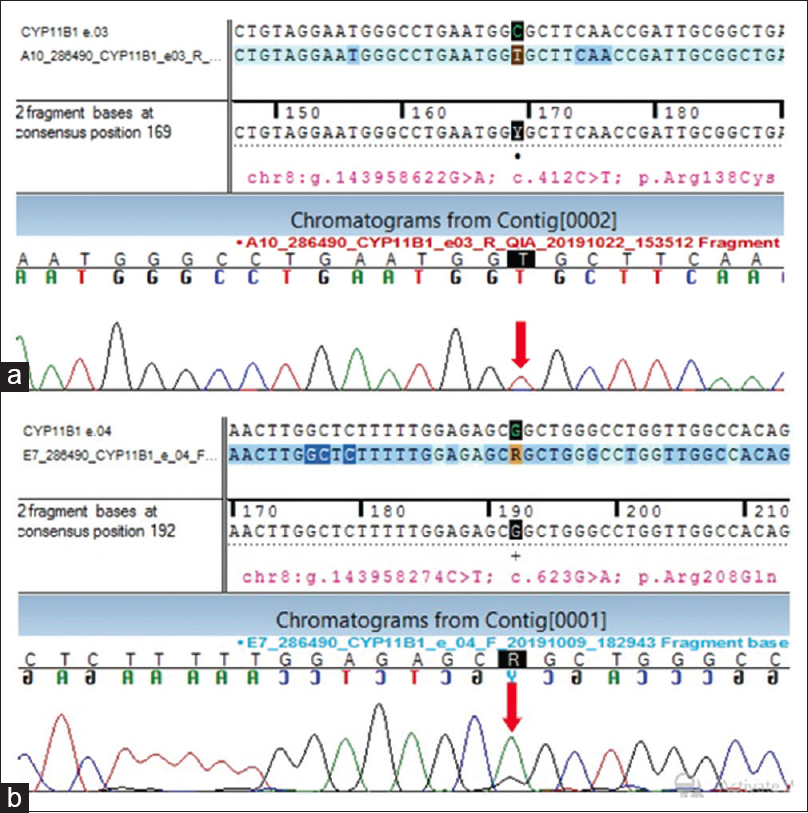
- (a) Sequence chromatogram and alignment to the reference sequence showing the variation in exon 3 of the CYP11B1 gene (chr8:g. 143958622G>A; c.412C>T; p.Arg138Cys) detected in homozygous condition. (b) Sequence chromatogram and alignment to the reference sequence showing the variation in exon 4 of the CYP11B1gene (chr8:g.143958274C>T; c. 623G>A; p.Arg208Gln) detected in homozygous condition
Discussion
CAH is a group of autosomal recessive disorders caused by an inherited defect in cortisol biosynthesis. 11-β hydroxylase deficiency (11β-OHD) accounts for 7% of all CAH, with an incidence of 1: 100,000 live births.[12] This condition occurs as a result of mutations in the CYP11B1 gene on chromosome 8q22. In classical 11OHD, mutations of the CYP11B gene results in decreased conversion of 11-deoxycortisol to cortisol, leading to elevated plasma ACTH levels and overproduction of steroid precursors. This results in virilisation of the female external genitalia and precocious pseudopuberty in both sexes, due to increased androgenic hormones, DHEA, and androstenedione. Also, the same enzyme CYP11B1 catalyzes the conversion of 11-deoxycorticosterone to corticosterone. Therefore, this deficiency of 11-β hydroxylase will result in elevated levels of 11-deoxycorticosterone, a potent mineralocorticoid manifesting as hypertension, while suppressing the renin secretion. Interestingly, the timing and appearance, of hypertension and other clinical features seems variable and based on this, classical and non-classical forms of 11OHD exists.[3]
The presentation in our patient was non-classical. On clinical examination, he was neither tall nor did he have any signs of hyperpigmentation due to raised ACTH. Also, he had normal cortisol levels. Nevertheless, on ACTH stimulation test, we were able to demonstrate very high levels of 11-deoxycortisol that could not be converted to cortisol, in the presence of 11-β hydroxylase deficiency. Also, studies have revealed that around 51 different mutations have been reported in about 100 patients with 11OHD.[4] In Moroccan Jews, in whom the condition has been more prevalent, the most common mutation was p.R448H (Arginine substituted for Histidine at codon 448).[5] But, our patient had two missense mutations that resulted in amino acid substitution of Cysteine for Arginine (p.Arg138Cys) at codon 138 and Glutamine for Arginine at codon 208, which might prove as a novel mutation that has to be screened for, in Indian population.
Treatment includes replacement glucocorticoid therapy that is, administering the largest dose of hydrocortisone at night. This is done to suppress the physiological early morning secretion of higher levels of ACTH, and also to reduce DOC secretion and normalize renin activity and corrects hypokalemia.[67] Our patient was initiated on tablet hydrocortisone 5 mg in the morning and 10 mg in the night. To provide holistic treatment, in view of long-term requirement of steroids, after taking his baseline bone mineral density, he was also started on Vitamin D and calcium supplements to prevent osteoporosis secondary to long term steroid therapy. He is currently on out-patient basis follow-ups with well-controlled blood pressure.
Conclusion
Diagnosis of CAH due to 11-β Hydroxylase deficiency, as a secondary cause of hypertension requires high index of suspicion, given that there is poor correlation between the genotype and phenotype of this condition. On contrary to use of conventional anti-hypertensives, steroids play a key role in the management of hypertension and hypokalemia, seen in this condition. We have described two novel mutations in our patient who has atypical presentation of congenital adrenal hyperplasia due to 11-β Hydroxylase deficiency, highlighting the need for genetic analyses in evaluation of hypertension during the current era of precision medicine.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
I thank Dr. Viknesh Prabu Anbalagan, Consultant, Viknesh Hospital, Cuddalore for his help in editing the manuscript.
References
- The incidence of congenital adrenal hyperplasia in Switzerland – A survey of patients born in 1960 to 1974. Helv Paediatr Acta. 1980;35:5-11.
- [Google Scholar]
- Steroid 11 beta- hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol Metab. 2008;19(3):96-99.
- [Google Scholar]
- Donor splice mutation in the 11beta-hydroxylase (CypllB1) gene resulting in sex reversal: A case report and review of the literature. J Pediatr Endocrinol Metab. 2006;19:1267-82.
- [Google Scholar]
- A mutation in CYP11B1 (Arg-448–His) associated with steroid 11 beta-hydroxylase deficiency in Jews of Moroccan origin. J Clin Invest. 1991;87:1664-7.
- [Google Scholar]
- Congenital adrenal hyperplasia: Not really a zebra. Am Fam Physician. 1999;59:1190-6, 1172.
- [Google Scholar]
- Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab. 1971;32:266-84.
- [Google Scholar]




