

Translate this page into:
IgG4 related interstitial nephritis: A case report and review of literature
Address for correspondence: Dr. N. Gopalakrishnan, Department of Nephrology, Madras Medical College and Rajiv Gandhi Government General Hospital, Chennai - 600 003, India. E-mail: srigola751@yahoo.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
IgG4 interstitial nephritis is a recently described entity. A middle-aged gentleman with bilateral parotid enlargement, hepatosplenomegaly and generalized lymphadenopathy was referred to us for evaluation of renal failure. He had trace proteinuria and large kidneys. Kidney biopsy revealed interstitial nephritis with characteristic storiform fibrosis. Immunohistochemistry demonstrated intense staining for IgG4-secreting plasma cells in the interstitium.
Keywords
IgG4
interstitial nephritis
storiform fibrosis

Introduction
IgG4-related disease (IgG4 RD) is a recently recognized novel systemic disease characterized by dense infiltration of affected Organ(s) with lymphoplasmacytic cells and fibrosclerosis. Clinical presentation depends on the organ involved, the prototype being auto immune pancreatitis. Salivary gland involvement is another major presentation. The commonest renal involvement in IgG4RD manifests as interstitial nephritis. Significant presence of IgG4 positive plasma cells and characteristic storiform fibrosis are the histopathological hallmarks of the disease. Elevated serum IgG4 levels (often, not always) and dramatic response to steroids are the other characteristic features.
Case Report
A 39-year-old man was referred for evaluation of renal failure. He complained of weight loss of about 8 kg and intermittent fever during the previous 3 months. There was no history of dry mouth or eyes. Examination revealed cervical, axillary and inguinal lymphadenopathy, bilateral parotid enlargement, and hepatosplenomegaly. There was trace ankle edema. Blood pressure was 140/90 mmHg. Examination of heart, lung, and nervous system revealed no abnormalities. Investigations: Urine analysis-trace proteinuria, spot urine protein-creatinine ratio: 0.53, no active sediments. Blood: Hemoglobin 11.8 g/dl; Total count 7800/mm3 (polymorphs 64%, lymphocytes 27%, monocytes 2%, eosinophils 7%), erythrocyte sedimentation rate 64 mm-1st h, peripheral smear revealed no abnormal immature white blood cells. Urea 56 mg/dl; creatinine 2.4 mg/dl; sodium 134 mEq/L, potassium 3.8 mEq/L, total serum protein 8.6 g/dl, albumin 3 g/dl, globulin 5.6 g/dl, serum transaminases were within the normal limits. Serology for HIV, HBsAg, and anti-HCV antibody were negative. Ultrasonogram of abdomen revealed large kidneys (right-18.2 × 7.2 cm; left-18.4 × 7.4 cm). CT abdomen confirmed the ultrasound findings, and no abnormalities were noted in pancreas. Renal biopsy: light microscopic examination revealed massive interstitial infiltration of lymphocytes, plasma cells, and a significant number of eosinophils with extensive fibrosis and tubular atrophy. Glomeruli were normal. Immunofluorescence for IgG showed intense positivity in the capillary walls. Stains for IgM, IgA, C3, C4, C1q, Kappa, and Lambda light chains were negative. Immunohistochemical stain for IgG4 was diffusely positive with most of the cells showing positivity (more than 50 cells/hpf) [Figures 1–3].
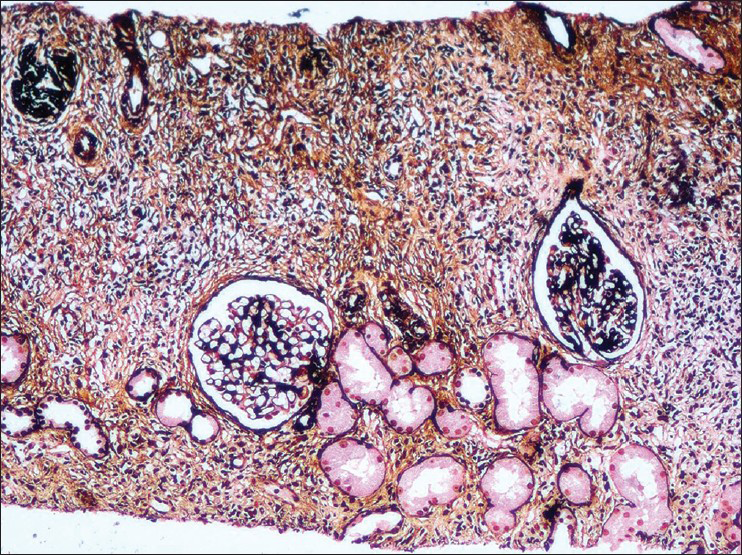
- A dense inflammatory infiltrate along with storiform fibrosis which destroys the interstitium. There is global glomerulosclerosis and periglomerular fibrosis. silver methenamine, ×100
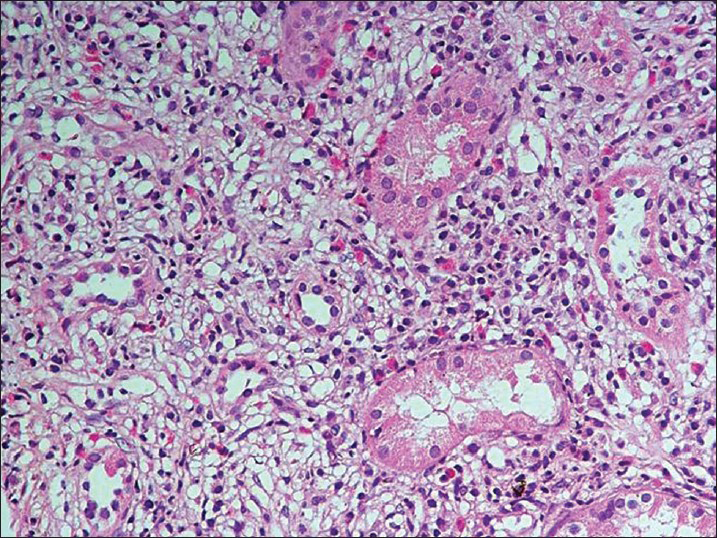
- The tubules are separated out by fibrous connective tissue and an inflammatory infiltrate composed of lymphocytes
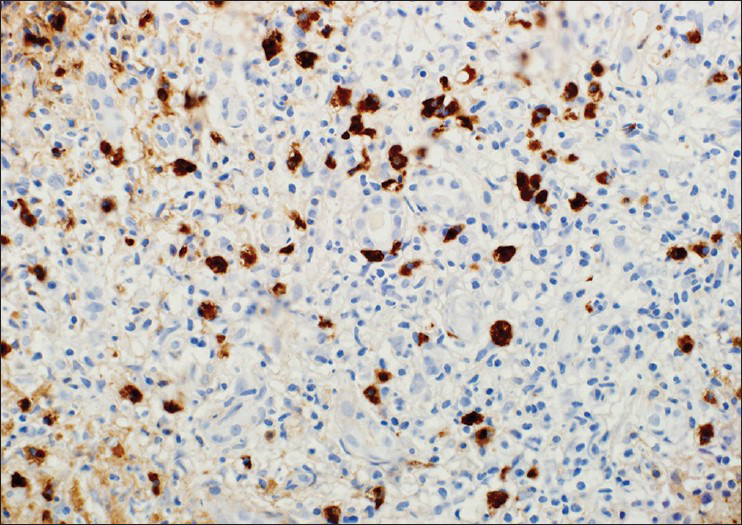
- IgG4 immunostaining shows more than 50IgG4 positive plasmacells per high powerfield
Discussion
IgG4 related disease is a multi-system disorder, the hallmark of which is infiltration of the involved organs by IgG4-secreting plasma cells with ensuing fibrosclerosis. IgG4-related disease was first described by Yoshida and Hamano, et al., in a patient with auto immune pancreatitis in 2001,[12] and subsequently, similar lesions were identified in almost all organs. At present, many inflammatory conditions are encompassed under the spectrum of IgG4-related disease including sclerosing cholangitis, sialadenitis, Riedel's thyroiditis, dacryoadenitis (Mikulicz disease), interstitial pneumonitis, idiopathic retroperitoneal fibrosis, periaortitis etc.
IgG4 is an unusual molecule with unique features. It is the rarest of the IgG subclasses to be expressed in serum accounting for only 3-6% of IgG levels. IgG4 has weaker inter-chain disulfide bridges so that immunoglobulin half molecules composed of one heavy chain and light chain dissociate from each other. Once they dissociate, an IgG4 molecule reassociates with another half molecule with specificity for a different antigen. Due to this “half-exchange antibody reaction,” IgG4 can neither fix complement nor form immune complex.[3]
The exact pathogenesis of IgG4RD is unknown. Autoimmunity and infective agents are hypothesized to be the immunological triggers. There is activation of T helper 2 cells (Th2) and subsequently of T regulatory cells (Tregs). Th2 cytokines IL-4, IL-5, and IL-13 result in eosinophilia and IgG4 class switch. IL-10 and TGF-β are secreted by Tregs. IL-10 favors differentiation of IgG4 class-switched B lymphocytes into plasma cells and TGF-β mediates fibrosis. Massive infiltration of lymphocytes and plasma cells results in tumorous swelling of the affected organ.[4]
Clinical features depend upon the organ involved. There are many common features in patients with IgG4-related disease irrespective of site of involvement viz., predominance in middle-aged to elderly men, elevated serum IgG4 levels, abundant infiltration of involved organ with IgG4- positive plasma cells, hypergammaglobulinemia, hypocomplementemia, and dramatic response to steroid therapy. The clinico-pathological features of IgG4-related disease have been extensively described in patients with auto immune pancreatitis. It usually affects middle-aged to elderly group, predominantly males (Male:Female ratio of 5:1). Thirty percent people can have extra pancreatic manifestations like sclerosing cholangitis, sclerosing sialadenitis, lymphadenopathy, and retroperitoneal fibrosis, which may occur during the course of illness or much later.
The proposed diagnostic criteria for this condition are shown in Table 1. Three patterns of renal lesions have been described in IgG4RD. (i) Diffuse tubulointerstitial nephritis with characteristic fibrosis and IgG4-positive plasma cells. IgG4+ cells are also detected in other pathologies as well like pauci-immune necrotizing and crescentic glomerulonephritis, chronic pyelonephritis[5] etc., Hence, quantitation of IgG4+ cells is important. Current consensus is that presence of >10 cells/hpf is diagnostic of IgG4RD;(ii) focal lymphoplasmacytic infiltration giving rise to tumor-like masses mimicking renal cell carcinoma;(iii) hydronephrosis secondary to retroperitoneal fibrosis.[6] Seventy to eighty percent patients with idiopathic membranous nephropathy have IgG4 auto antibodies against a protein found in glomerular podocyte called M-type phospholipase A2 receptor (PLA2 R), raising a question of both entities sharing a common antigen.[7] But studies by Khosroshahi, et al,[8] showed that none of the 28 patients with IgG4 RD had PLA2R antibodies.

Our patient fulfills the mandatory criteria of presence of more than 10 IgG4 positive plasma cells per high power field. He had large kidneys, hypergammaglobulinemia, eosinophilia, bilateral submandibular, and parotid gland enlargement, axillary and cervical lymphadenopathy. Due to logistic reasons we could not do serum IgG4 levels, which may be elevated in 60-70% patients.
In a retrospective study of 153 patients with suspected IgG4-related disease from Japan, 23 patients (15%) were identified with IgG4-related tubulointerstitial nephritis all but one of whom (96% of TIN patients) exhibited involvement of other organs. These extra renal manifestations included sialadenitis (83%), lymphadenopathy (44%), autoimmune pancreatitis (39%), dacryoadenitis (30%), lung lesions (26%), and others in individual patients. 26% patients had associated glomerular lesions (3-undefined mesangio-proliferative glomerulonephritis, 2-membranous nephropathy, 1-IgA nephropathy).[10]
One important differential diagnosis of IgG4 RSD is Sjogren's syndrome. Male preponderance, relatively mild forms of xerophthalmia and xerostomia despite enlargement of their lacrimal and salivary glands, characteristic infiltrates with IgG4 positive plasma cells, good response to steroids, and elevation of IgG4 and IgE levels favor the diagnosis of IgG4-related disease. The natural history of IgG4 RD is still unclear. Even though it has overall good prognosis, there are several case reports of IgG4-related disease being associated with malignancy of pancreas, salivary glands, and ocular adnexal lymphomas.
Even though no randomized control trials have been conducted in IgG4RD, aggressive treatment with corticosteroids is indicated when vital organs are involved to prevent severe organ dysfunction. Glucocorticoids are the first line of therapy. Consensus group from Japan recommends starting steroids at dose 0.6 mg/kg for 2-4 weeks followed by tapering to 5 mg by 3-6 months and maintaining at 2.5-5 mg for 3 years. Azathioprine, mycophenolatemofetyl, and methotrexate can be used as steroid-sparing drugs or to maintain remission.[11] A major determinant of clinical responsiveness depends upon the degree of fibrosis within the affected organ.
Conclusion
As the concept of IgG4 RD is proposed recently, it remains largely unrecognized in regular clinical practice. It should be considered in differential diagnosis of tubulo-interstitial nephritis, particularly in elderly men with the known extra renal manifestations. Prompt diagnosis would be rewarding given the dramatic response to steroids in the early stages of the disease. To the best of our knowledge, ours is the first case-report of IgG4RD from India.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Chronic pancreatitis caused by auto immune abnormality. Proposal of concept autoimmune pancreatitis. Dig Dis Sci. 1995;40:1561-8.
- [Google Scholar]
- High serum IgG4 concentrations in sclerosing pancreatitis. N Engl J Med. 2001;344:732-8.
- [Google Scholar]
- Significance of IgG4 immuno staining in plasma cell rich tubulointerstitial cell nephritis. USCAP 2011 abstract 1484
- [Google Scholar]
- A case of IgG4 related systemic disease with markedly enlarged kidneys. NDT plus. 2009;2:233-5.
- [Google Scholar]
- IgG4-related tubulointerstitial nephritis with membranous nephropathy. Am J Kidney Dis. 2011;58:320-4.
- [Google Scholar]
- IgG4-related disease is not associated with antibody to the phospholipase A2 receptor. Int J Rheumatol 2012 2012 139409
- [Google Scholar]
- Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15:615-26.
- [Google Scholar]
- Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78:1016-23.
- [Google Scholar]
- Japanese consensus guidelines for management of auto immune pancreatitis. J Gastroenterol. 2010;45:471-7.
- [Google Scholar]




