

Translate this page into:
Proliferative Glomerulonephritis with Monoclonal Immunoglobulins in a Child with Recurrence in the Allograft
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) is among the spectrum of monoclonal protein-associated renal diseases, with only about 15 case reports in children. We report a 7-year-old boy with biopsy-proven crescentic PGNMID who progressed to end-stage renal disease within a few months of presentation. He then received a renal transplant with his grandmother as a donor. Proteinuria was detected at 27 months post-transplant and an allograft biopsy revealed a recurrent disease.
Keywords
Monoclonal gammopathy
PGNMID
recurrence

Introduction
The first series of cases of proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) was described by Nasr et al. in 2004.[1] This was characterized by immunoglobulin deposits with a single heavy and single light chain with no substructure mimicking immune-complex-mediated glomerulonephritis. In the pediatric age group, this entity is distinctly rare, with only 15 cases being reported so far.[2-4] We report one such unusual case in a 7-year-old boy with progression to end-stage renal disease with recurrence in the graft.
Case Report
A 7-year-old boy, born to non-consanguineous parents and with no significant illness in the past, presented with worsening pedal edema and facial puffiness over 2 weeks. The patient reported a history of chickenpox 1 month prior to the onset of edema. He did not have any gross hematuria or dysuria. His vitals were stable with normal blood pressure. General and systemic examinations were normal except for the presence of pitting bilateral pedal edema and facial puffiness. Laboratory evaluation and physical examination helped to establish the diagnosis of nephrotic syndrome. Labs at presentation were urine protein 3+, RBC 20-25/hpf, WBC 12–14/hpf, serum creatinine was 0.99 mg/dl, serum albumin was 2.2 gm/dL, and serum cholesterol was 182 mg/dl. C3 level was low at 0.603 gm/l (normal range: 0.9–1.8 gm/l), and C4 was normal at 0.109 gm/l (normal range: 0.1–0.4 gm/l). Renal biopsy was done in view of elevated serum creatinine at presentation.
The biopsy showed, on light microscopy, 18 glomeruli with a membrano-proliferative glomerulonephritis (MPGN) pattern of injury, five of them with crescents. There was mild tubular injury and mild interstitial inflammation. Tubular atrophy and interstitial fibrosis were not noted, and the blood vessels were unremarkable. Tissue submitted for IF study had a single glomerulus in which the pattern of immune deposits was not clearly discernible. The tissue taken for EM study showed no glomeruli.
The child was pulsed with IV methylprednisolone 20 mg/kg body weight for five doses and thereafter continued on oral prednisolone 1 mg/kg body weight. He was started on IV cyclophosphamide at 500 mg/m2 per dose. Six doses of IV cyclophosphamide were planned at 3–4 weekly dosing intervals. After two doses of cyclophosphamide, the child had a partial response. However, soon after the third dose of cyclophosphamide, the child developed lower respiratory tract infection, and there was a rapid progression of the disease with oligo-anuria and worsening edema, development of hypertension, worsening of the serum creatinine to 3.2 mg/dL, and increase in proteinuria to a urine protein: creatinine ratio of 9.8. The child was admitted in the pediatric ICU for hypertensive emergency. The course in the ICU was very stormy, with development of posterior reversible encephalopathy syndrome and worsening of oligo-anuric renal failure. He was managed with multiple antihypertensives and started on hemodialysis. He also underwent five cycles of plasmapheresis. Despite all the measures, the child continued to have dialysis dependent renal failure and at this point after stabilization, a second renal biopsy was done within a month of acute progression of the illness.
The repeat biopsy showed 14 glomeruli, all with fibrocellular and cellular crescents. The underlying tuft had an uniform MPGN pattern of injury with mesangial and endocapillary hypercellularity and a diffuse thickening and duplication of the basement membrane [Figure 1]. There was mild tubular atrophy and interstitial fibrosis involving 25% of the cortex sampled with mild interstitial inflammation. The blood vessels were unremarkable. Direct immunofluorescence study showed strong peripheral and mesangial deposits of IgG, C3c, and C1q. There was kappa light chain restriction [Figure 2]. The ultrastructural studies showed electron dense deposits with no substructure in the subendothelial and mesangial areas with occasional subepithelial deposits. There was diffuse foot process effacement [Figure 3]. With these findings, a diagnosis of PGNMID with IgGk deposits with crescents and mild tubular atrophy was made.

- The left panel shows glomeruli with crescents (Jones silver 100×). The right panel shows a glomerulus with mesangial widening and hypercellularity with a thickening of the basement membrane and an overlying cellular crescent (HE 400×)
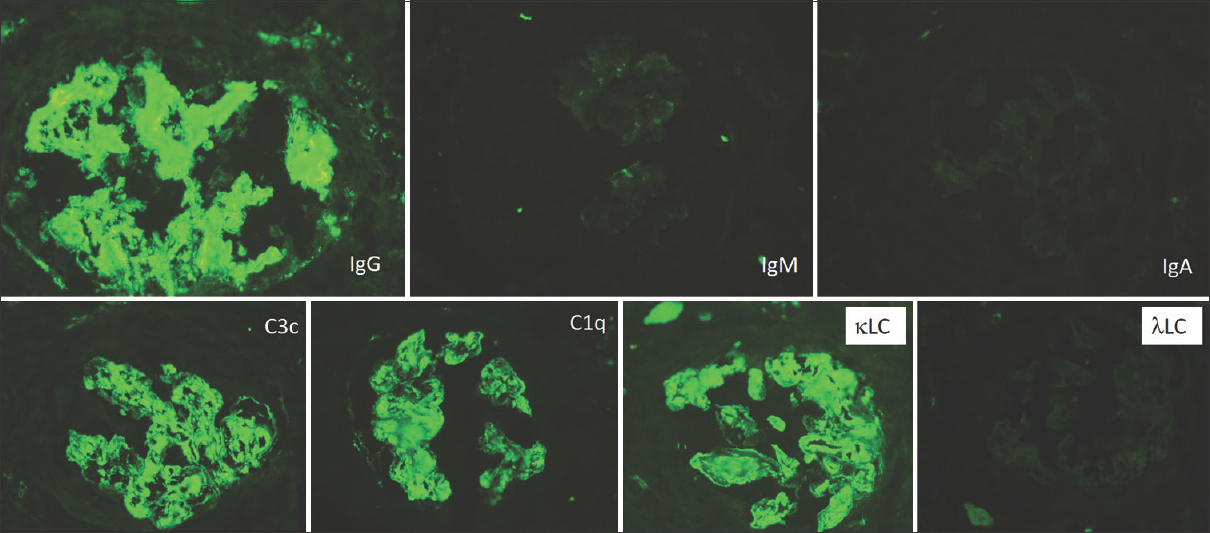
- Composite immunofluorescence panel showing strong positivity for IgG, C3, and C1q as prominent mesangial smudgy deposits and around the capillary loops. There is light chain restriction with strong κ light chains and negative λ light chains (FITC-labeled antibodies 200×)
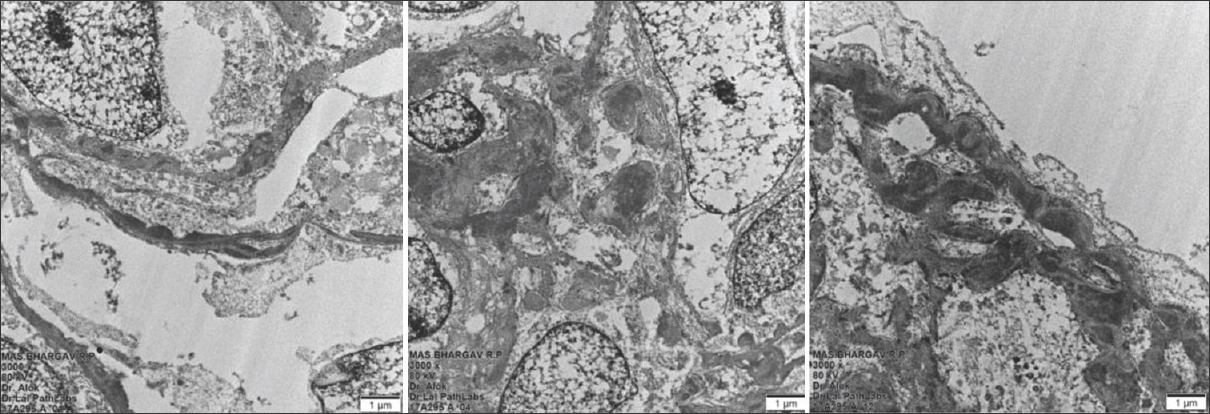
- Electron microscopy showing mesangial electron-dense granular deposits in the subendothelial location (left panel) and mesangial location (middle panel). The right panel shows subendothelial deposits and diffuse foot process effacement (3000×)
Bone marrow examination, serum and urine electrophoresis, and free light chain assays did not show any evidence of a monoclonal paraproteinemia. The child was continued on prednisolone but continued to be dialysis-dependent. The prednisolone eventually was tapered and stopped. Twenty-one months after the second biopsy, a transplant was planned with the maternal grandmother as the donor. One dose of rituximab at 375 mg/m2 body weight was given prior to the transplant. The transplant itself was uneventful and the post-operative recovery of renal function was as expected. He was maintained on triple-drug immunosuppression with steroids, tacrolimus, and mycophenolate.
The boy was on regular follow-up and was doing well till 22 months post-transplant, when there was an irregularity in his treatment because of the Covid pandemic causing anxiety and disruption in the family and he was lost to follow-up for over 6 months thereafter. There was no recorded covid positivity in the patient or any of the family members. Routine investigations done after about 6 months period of irregular treatment and follow-up revealed proteinuria with microscopic hematuria. His serum creatinine was elevated to 1.2 mg/dl, serum albumin was normal at 3 gm/dl, and tacrolimus trough level was low. Clinically, he was stable with no edema or hypertension. After optimizing the treatment, the serum creatinine decreased to 0.8 mg/dL; however, the child continued to have significant proteinuria of 1.3 g/24 h and microscopic hematuria. Suspecting disease recurrence or early graft rejection, an allograft renal biopsy was done.
The third renal biopsy, which was an allograft biopsy, done about 30 months after renal transplant, showed 10 glomeruli with a mild mesangial proliferation with mesangial widening, an irregular thickening, and duplication of the basement membrane. There were no crescents. The tubulointerstitial and vascular compartments were unremarkable. There was no evidence of rejection [Figure 4]. The direct immunofluorescence study revealed a patten of deposits identical to that in the native kidney with IgGk deposits [Figure 5]. This time, IgG isotype stains were also done, and the deposits were of the IgG3 subtype [Figure 6]. C4d immune stains showed no positivity in the peritubular capillary walls. There was peripheral and mesangial positivity for C4d in the glomeruli.
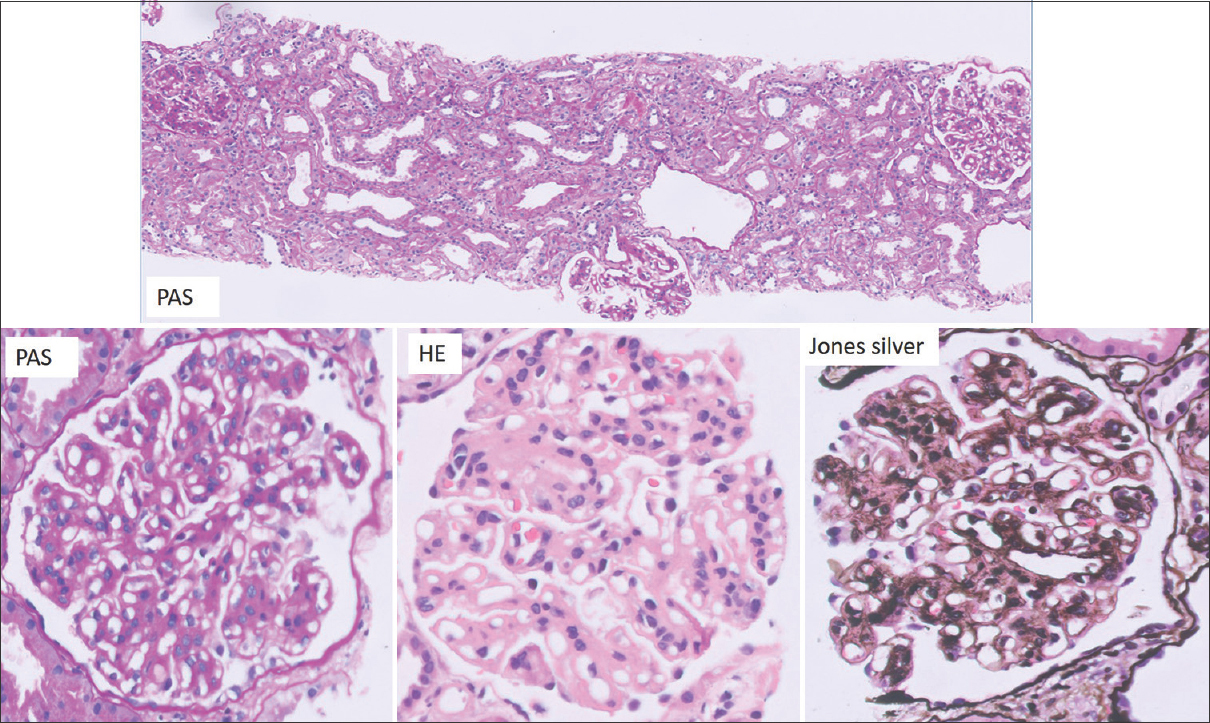
- The top panel shows two hypercellular glomeruli with an unremarkable tubule-interstitium (PAS 100×). The bottom panel shows the glomerulus with a mesangial hypercellularity and an irregular thickening of the basement membrane with focal double contours (400×)
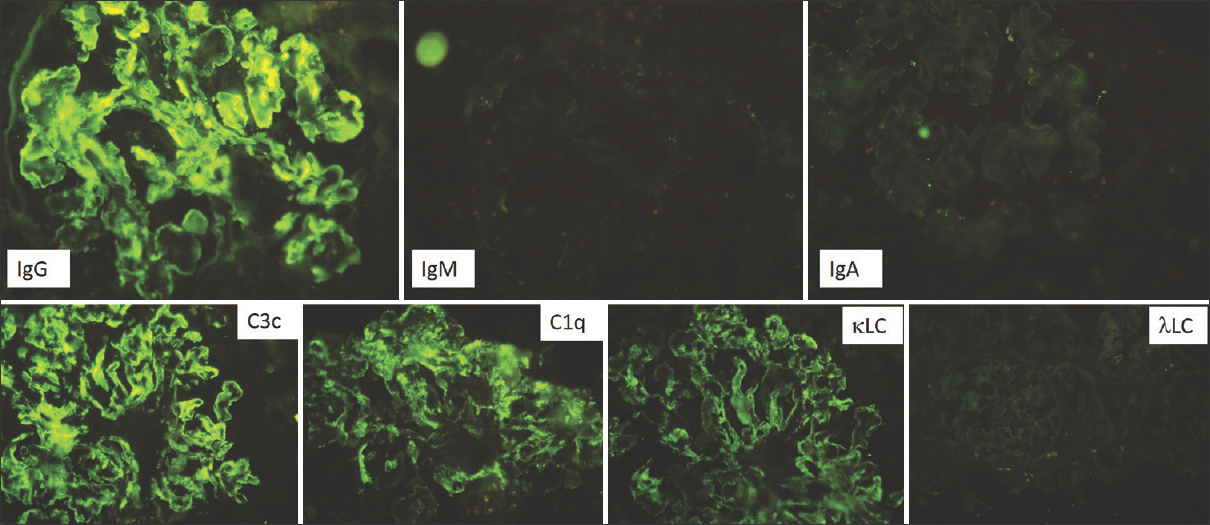
- Immunofluorescence of the allograft biopsy showing strong staining for IgG, C3c, and C1q with kappa light chain restriction, similar to that seen in the native kidney (FITC- labeled antibodies 200×)
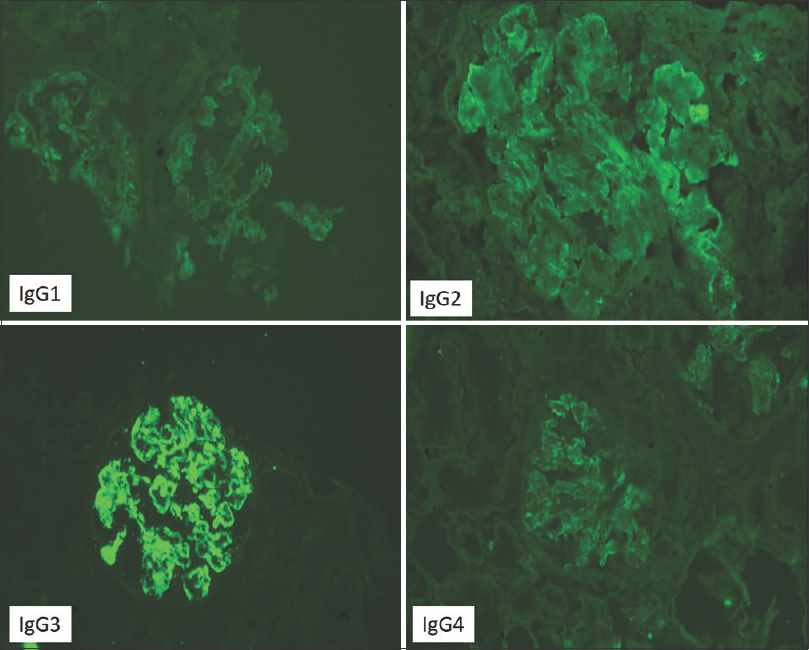
- IgG isotype staining showing positivity for IgG3 and negative for IgG1, IgG2 and IgG4.I (FITC-labeled antibodies 200×)
The diagnosis was concluded as recurrent PGNMID IgG3k-type with no chronicity or evidence of rejection. A single dose of rituximab at 375 mg/m2 was given and the same immunosuppression continued. At 2 months follow-up, there was no increase in proteinuria, and the serum creatinine was maintained at 0.8 mg/dL
Discussion
PGNMID is a rare disease among the spectrum of monoclonal gammopathy of renal significance (MGRS) and was first described by Nasr et al. in 2004.[4,5] This is a disease predominantly of adults, more common in women manifesting as nephrotic syndrome with hematuria and varying degrees of renal failure. On light microscopy, a MPGN pattern of injury is the most common with mesangioproliferative, endocapillary proliferative, and rarely a membranous pattern also being recorded. On immunofluorescence, there is usually deposition of a single gamma heavy chain subclass and a single light chain, with IgG3k being the most common. The deposits are almost always accompanied by deposits of C3c, with C1q also seen in up to 60%. The deposits on ultrastructure are primarily granular, resembling ordinary immune-complex type deposits. More recently, variants with only light chain deposits have been described, with an underlying plasma cell clone detectable in the majority and with a response to anti-plasma cell therapy.[6]
Only about a third of these cases have a demonstrable circulating monoclonal paraprotein, and only a single case in the large series by Nasr et al.[5] had multiple myeloma. The prognosis of this disease is variable, with nearly a quarter of these patients progressing to end-stage renal disease despite immunosuppression.
PGNMID in children is rare, with the first case being reported only in 2018. Subsequently, there have been two small case series of five and nine cases.[1-3] Nephrotic syndrome with hematuria is again the most common form of presentation. The MPGN pattern of injury is the most common, with the deposits being IgGk in most, though in the series by Miller et al.,[3] IgG3λ was seen in seven of the nine patients. Three of the nine biopsies in this series had crescents. An underlying monoclonal paraprotein was not detected in any of the children. The condition was found to be slowly progressive in the majority despite immunosuppression.
The pathogenesis of PGNMID is still not fully understood. In the pediatric case series by Miller et al.,[3] half the children had a preceding infection or low complement levels. Two of the three children tested demonstrated genetic mutations of the alternate complement pathway. It is postulated that intrinsic or extrinsic antigens could trigger a polyclonal or oligoclonal immune response, and IgG3 antibodies thus generated are notably nephrotoxic and also activate complement. These responses may be amplified in individuals with an underlying complement or immune dysregulation.[4] There have been two case reports of adults with PGNMID evolving from a C3 glomerulopathy, with both having an underlying CFH mutation.[7,8] It is interesting that our child had an episode of chickenpox 1 month prior to the onset of the nephrotic illness; this could have been the trigger for the immune response. Genetic testing for the dysfunction of the alternate complement pathway was not done as these tests were not available.
In adult patients of PGNMID followed post-transplant, there is a high rate of recurrence.[9] In a cohort of 26 patients, 89% had a recurrence with 5.5 months being the median time to recurrence. MPGN pattern with IgG3 subclass deposits was the most common finding in allograft biopsies. Forty-four percent lost their graft within a mean of 36 months from the diagnosis. In another 44%, there was a relapse despite initial response to immunosuppression.[10] In the pediatric series by Miller et al.,[3] six of the nine children developed kidney failure, and four received allografts. A recurrence was recorded in three of them, with one patient having a recurrence in three separate kidney allografts over a 20-year period.
In conclusion, it can be said that PGNMID in children is rare and can present as a crescentic GN. Progression of the disease to ESRD is not uncommon and there is a high rate of recurrence post-transplant. This case also highlights the importance of use of light chain antisera in the immunofluorescence panel in all pediatric renal biopsies, without which the diagnosis would not have been possible.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Proliferative glomerulonephritis with monoclonal IgG deposits: A-distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65:85-96.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal immunoglobulin G lambda deposits: Report of the first pediatric case. Case Rep Nephrol Dial. 2018;24(8):70-5.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal IgG deposits in children and young adults. Pediatr Nephrol. 2018;33:1531-8.
- [Google Scholar]
- Progression of proliferative glomerulonephritis with monoclonal IgG deposits in pediatric patients. Pediatr Nephrol. 2021;36:927-37.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal IgG deposits. J-Am Soc Nephrol. 2009;20:2055-64.
- [Google Scholar]
- Light chain only variant of proliferative glomerulonephritis with monoclonal immunoglobulin deposits is associated with a high detection rate of the pathogenic plasma cell clone. Kidney Int. 2020;97:589-601.
- [Google Scholar]
- Post-infectious proliferative glomerulonephritis with monoclonal immunoglobulin G deposits associated with complement factor H mutation. Intern Med. 2017;56:811-7.
- [Google Scholar]
- A-case of switch from C3 glomerulonephritis to proliferative glomerulonephritis with monoclonal IgG deposits. Ann Clin Lab Sci. 2018;48:528-33.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol. 2011;6:122-32.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal immunoglobulin G deposits is associated with high rate of early recurrence in the allograft. Kidney Int. 2018;94:159-69.
- [Google Scholar]




