

Translate this page into:
Rare Association of Waardenburg Syndrome with Minimal Change Disease
Address for correspondence: Dr. S. B. Raju, Department of Nephrology, Nizam's Institute of Medical Sciences, Hyderabad - 500 082, Telangana, India. E-mail: sreebhushan@hotmail.com
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Waardenburg syndrome (WS) is a rare genetic disorder characterized by varying degrees of hearing loss, pigmentary anomalies, and defects of other neural crest cell-derived structures. The association of WS with renal anomalies has been described in the literature. However, nephrotic syndrome is a very rare association with WS, and only one case has been reported in the literature. We report a case of WS2 associated with biopsy-proven nephrotic syndrome (minimal change disease).
Keywords
Minimal change disease
Waardenburg syndrome
white forelock

Introduction
Waardenburg syndrome (WS) is named after the Dutch Ophthalmologist Petrus Johannes Waardenburg. It was Van der Hoeve in 1916 who described deaf mutism in association with eye anomalies in a pair of 14-year-old monozygotic twin girls.[1] Petrus Johannes Waardenburg in 1947 first described a patient with hearing loss, dystopia canthorum (i.e., lateral displacement of the inner canthi of the eyes), and retinal pigmentary differences.[2] The condition described originally is now categorized as WS1. Since then, four subtypes (I to IV) with variable penetrance of different clinical features have been described.[3] We report a rare presentation of WS2 with nephrotic syndrome.
Case Report
A 19-year-old female presented with chief complaints of anasarca, exertional dyspnea, and decreased urine output for 1 month. She is born out of a nonconsanguineous marriage with uneventful perinatal history. Her family gives a history of congenital deafness with rest of the milestones attained as per age. The patient denies any history of febrile illness, dysuria, hematuria, bleeding manifestations, oral ulcers, hair loss, vomiting, abdominal pain, blurring of vision. She does not give any history of systemic illness or use of chronic medications. The patient had centrally placed white forelock (artificially dyed for cosmetic reasons). Her father and her siblings also have a white forelock with premature graying of hair [Figure 1].

- Pedigree chart showing autosomal dominant incomplete penetrance of Waardenburg syndrome in this patient's family
On systemic examination, she has distinctive facial abnormalities (high nasal bridge, synophrys, unusually rounded nasal tip, abnormal smoothness of the philtrum). Similar abnormal facial features were seen in her sister.
On ocular examination, her visual acuity, color vision, and intraocular pressure were within normal limits. Inner canthal distance, interpupillary distance, and outer canthal distance were 40, 75, and 130 mm, respectively, and Waardenburg index (W index) was 1.67. She has heterochromia iridis in the right eye. Lacrimal system was patent. Rest of the anterior segment was normal in both eyes. Dilated fundus examination showed tessellated hypopigmented fundus in the right eye and normal fundus in the left eye [Figure 2].
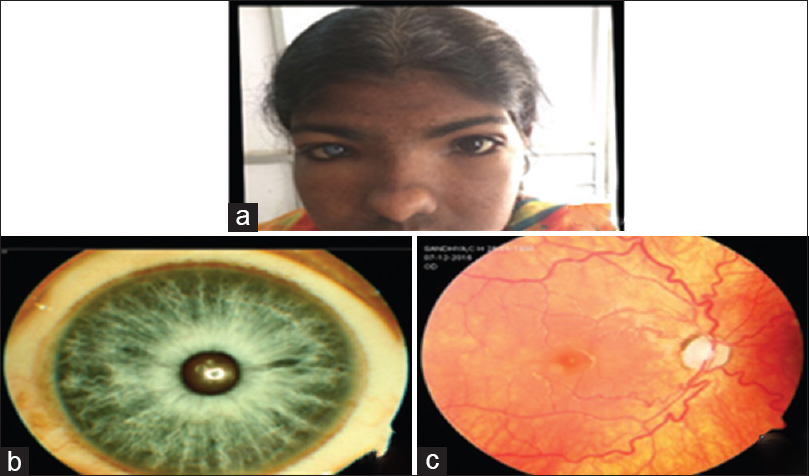
- (a) White forelock, abnormal facies; (b) heterochromia iridis; (c) tessellated hypopigmented fundus
Audiometry revealed profound bilateral sensory neural hearing loss.
She was further evaluated and her urine examination revealed nephrotic range proteinuria (8.6 g/day). Urine sediment was bland. Serum urea and creatinine levels were within normal limits. Antinuclear antibody, double-stranded DNA, and antineutrophil cytoplasmic antibody were negative. Serum copper and ceruloplasmin levels were within normal limits. Renal biopsy was done and light microscopy showed 12 glomeruli without any morphological changes and immunofluorescence was negative for immunoglobulin deposits, which were suggestive of minimal change disease (MCD). The electron microscopy revealed diffuse effacement of visceral epithelial cell foot processes; glomerular basement membrane (GBM) thickness varies from 268.4 to 339.5 nm, with a mean of 295.8 nm, which is normal for age. No electron dense or organized deposits were seen in mesangial areas. The ultrastructural features indicate a primary podocytopathy [Figure 3]. She was started on prednisolone (1 mg/kg/day) and remission was observed within 2 weeks of initiation of treatment. Proteinuria after 2 months of initiation of treatment was 100 mg/day, and she was asymptomatic.
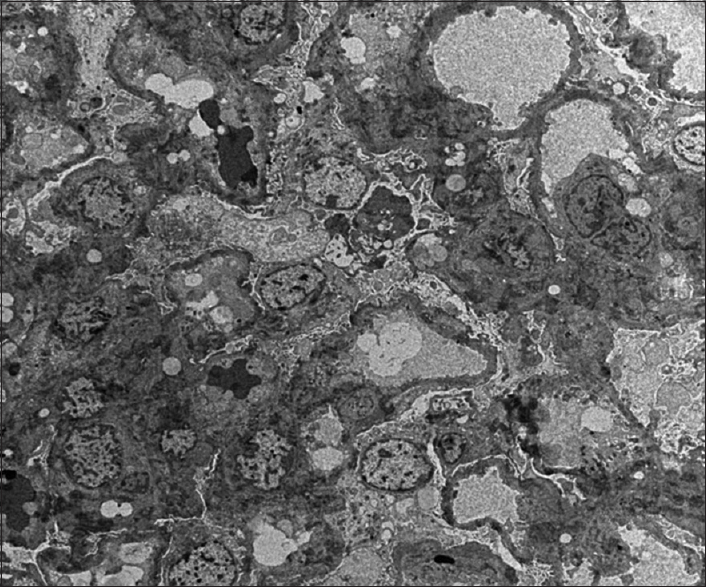
- Electron microscopy image showing diffuse effacement of visceral epithelial cell foot processes. No electron dense or organized deposits are seen in mesangial areas of glomerular basement membrane. The ultrastructural features indicate a primary podocytopathy
Discussion
WS is a genetic disorder that may be evident at birth. The range and severity of associated symptoms and findings may vary greatly from case to case. There are five major and five minor criteria for WS. Major criteria include sensory neural hearing loss, iris pigmentary abnormality, hair hypopigmentation, dystopia canthorum, and first-degree relative previously diagnosed with WS. Minor criteria include skin hypopigmentation, medial eyebrow flare (synophrys), broad nasal root, hypoplasia, alae nasi, and premature graying of the hair.
According to the diagnostic criteria proposed by the Waardenburg consortium, a person must have two major or one major plus two minor criteria to be diagnosed as WS1.[4] WS2 lacks dystopia canthorum. WS3 is similar to WS1, but it is associated with upper limb defects. WS3 is the rarest form of WS. WS4 is associated with Hirschsprung's disease. The present case had three major criteria and five minor criteria of WS. Dystopia canthorum is diagnosed by W index. The measurements necessary to calculate the W index (in mm) are inner canthal distance (a), interpupillary distance (b), and outer canthal distance (c).
Calculate X = (2a − (0.2119c + 3.909))/c
Calculate Y = (2a − (0.2479b + 3.909))/b
Calculate W = X + Y + a/b
W index >1.95 confirms dystopia canthorum, which is a typical feature of WS1 and it is typically absent in WS2. W index in this patient was 1.67 confirming the case as Type 2.
WS1 is caused by mutations in the PAX3 gene located on chromosome band 2q35WS. Mutations in WS1 and WS2 may be inherited in an autosomal dominant pattern or may be de novo. Abnormalities in the PAX3 gene account for most of the WS1 and WS3 patients. Microphthalmia-associated transcription factor gene abnormality is responsible for WS2.[56] Mutations in PAX3 have also been found in patients with a WS3 phenotype.[7] WS4 is caused by homozygous mutations in either the endothelin 3 gene or the endothelin receptor type B gene. Heterozygous mutations in either of these genes cause isolated Hirschsprung's disease.[891011] SOX10 mutations are associated with a more severe phenotype known as PCWH, consisting of peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, WS, and Hirschsprung's disease.
MCD is an important cause of nephrotic syndrome and is characterized by massive proteinuria and hypoalbuminemia, resulting in edema and hypercholesterolemia. This case is a rare syndromic presentation.
There are few cases of renal anomalies associated with WS, which are reported in the literature. The first case of WS associated with duplication of renal collecting system was reported by Tiger and de Chaderevian in 1988.[12] Multicystic dysplastic kidney associated with WS1 was reported by Jankauskiene et al. in 1997.[13] Ekinci et al. published WS with right atrophic kidney and left double collecting system in 2005.[14] Pembegül et al. described the first association of WS1 with nephrotic syndrome in 2015.[15] We report this case of nephrotic syndrome in a female of WS with autosomal dominant inheritance with variable penetrance. The genetic analysis could not be performed in our case. The probable genetic association between WS and nephrotic syndrome needs further evaluation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We are thankful to Dr. Harika for helping us in preparing this manuscript.
References
- Abnorme Lange der Tranenrohrchen mit Ankyloblepharon. Klin Monbl Augenheilkd. 1916;56:232-8.
- [Google Scholar]
- A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet. 1951;3:195-253.
- [Google Scholar]
- Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: First report of the WS consortium. Am J Hum Genet. 1992;50:902-13.
- [Google Scholar]
- Mouse and hamster mutants as models for Waardenburg syndromes in humans. J Med Genet. 1990;27:618-26.
- [Google Scholar]
- Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8:251-5.
- [Google Scholar]
- Autosomal dominant inheritance of Klein-Waardenburg syndrome. Am J Med Genet. 1992;42:320-2.
- [Google Scholar]
- Amissense mutation of the endothelin-B receptor gene in multigenic hirschsprung's disease. Cell. 1994;79:1257-66.
- [Google Scholar]
- Endothelin receptor-mediated signaling in hirschsprung disease. Hum Mol Genet. 1996;5:303-7.
- [Google Scholar]
- Mutation of the endothelin-receptor B gene in Waardenburg-Hirschsprung disease. Hum Mol Genet. 1995;4:2407-9.
- [Google Scholar]
- Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome) Nat Genet. 1996;12:442-4.
- [Google Scholar]
- Piebaldism-Waardenburg syndrome: Histopathologic evidence for a neural crest syndrome. Am J Med Genet. 1988;31:679-88.
- [Google Scholar]
- Multicystic dysplastic kidney associated with Waardenburg syndrome type 1. Pediatr Nephrol. 1997;11:744-5.
- [Google Scholar]
- Waardenburg syndrome associated with bilateral renal anomaly. J Pediatr Surg. 2005;40:879-81.
- [Google Scholar]
- Waardenburg syndrome and nephrotic syndrome coexistence. SDU J Health Sci. 2015;6:41-2.
- [Google Scholar]




