

Translate this page into:
Renal Amyloidosis in a Child with Recessive Dystrophic Epidermolysis Bullosa Due to a Novel Variant in COL7A1 Gene
Corresponding author: Dr. Lesa Dawman, Department of Pediatrics, Room No. 5120, 5A, Advanced Pediatrics Centre, PGIMER, Chandigarh - 160 012, India. E-mail: lesadawman@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Daniel R, Dawman L, Nada R, Sekar A, Mahajan R, Tiewsoh K. Renal Amyloidosis in a Child with Recessive Dystrophic Epidermolysis Bullosa Due to a Novel Variant in COL7A1 Gene. Indian J Nephrol. 2024;34:400-3. doi: 10.4103/ijn.IJN_26_21
Abstract
Secondary amyloidosis may complicate inherited dermatoses, but recessive dystrophic epidermolysis bullosa (RDEB) complicated by renal amyloidosis is rare. We report a case of a 12-year-old male child with RDEB presenting with progressive generalized anasarca for 20 days. Kidney biopsy showed diffuse expansion of mesangial matrix by pale acellular Periodic Acid-Schiff (PAS)-negative amorphous material, which was congophilic on Congo red stain and gave apple green birefringence on polarization and extending along the glomerular basement membrane, suggestive of amyloidosis. Genetic analysis showed a compound heterozygous pathogenic variant in the COL7A1 gene with autosomal recessive inheritance.
Keywords
Amyloidosis
COL7A1 gene
Recessive dystrophic epidermolysis bullosa

Introduction
Epidermolysis bullosa (EB) is a rare inherited heterogenous group of skin disorders characterized by fragility of skin and mucosa with easy blister formation.1 The onset of symptoms is usually in infancy or early childhood. There are four main groups of EB according to the severity and extent of the disease: epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), dystrophic epidermolysis bullosa (DEB), and Kindler syndrome.1 DEB is caused due to a defect in type VII collagen gene and has an autosomal dominant or recessive inheritance.2 Recessive dystrophic epidermolysis bullosa (RDEB) is one of the severe forms of EB which has its onset within weeks after birth, and it typically has a more generalized involvement leading to scarring and contracture of hands, feet, and joints, corneal erosions, extensive mucosal involvement, esophageal strictures, dystrophy of the nails, pseudosyndactyly, scaring alopecia, enamel hypoplasia, microstomia, ankyloglossia, malnutrition, anemia, and growth retardation.3 Based on the presenting symptoms and family history of DEB, the diagnosis is suspected, and it is confirmed with either immunofluorescence antigen mapping of skin biopsy or a mutational analysis.4 Secondary amyloidosis is considered as a common and life-threatening complication of EB. In patients with RDEB, amyloidosis can involve the kidneys, liver, bowel, and respiratory system; however, the involvement of kidneys is rare and there are only a few case reports in literature.5-7
Case Report
A 12-year-old male child, born to nonconsanguineous parents, presented with complaints of generalized skin blisters and oral erosions since birth. There was history of trauma-induced excoriation of the skin. It gradually worsened to involve whole of his body in the past 4 months. He had been having difficulty in swallowing of solid and dry foods for the past 3 years. He was clinically diagnosed with RDEB and was being managed conservatively with wound care, nutritional supplements, antihistamines, and topical emollients. He was referred to us in view of swelling of face and around the eyes with frothuria of 20 days duration. It was gradually progressing with further involvement of lower limbs, trunk, and abdomen. There was no history of fever, cough, pain abdomen, decrease in urine output, or hematuria. He had a significant family history of one elder sibling loss. She was also diagnosed as a case of EB and had generalized skin lesions since birth; she succumbed to secondary infections at 8 years of age. In our index case, possibility of nephrotic syndrome with secondary amyloidosis was kept and was further investigated. On examination, he had widespread cutaneous erosions all over the body with generalized anasarca. At places, the erosions were healing with milia formation and scarring. There was nail dystrophy with contracture of bilateral elbows and corneal clouding [Figure 1]. His vitals were stable with normal anthropometry. On evaluation, his hemogram showed presence of anemia (hemoglobin- 9.0 g/dL, total leukocyte- 8.5 × 109/L, neutrophils- 64.2%, lymphocytes- 27.7%, monocytes- 8%, eosinophils- 0.1%, and platelets- 6.03 × 103/L). His metabolic profile showed serum creatinine- 0.26 mg/dL, total cholesterol- 284 mg/dL, total protein- 3.6 g/dL, and serum albumin- 0.9 g/dL. Serum amyloid A (SAA) level was 1160 mg/L (normal: <6.4 mg/L). The 24-h urine protein was 459.75 mg/m2/h. Urine examination revealed 4+ albumin and no red blood cells (RBCs), white blood cells (WBCs), casts, crystals, or sugar. Urine culture was sterile. Ultrasound of the abdomen was normal. Kidney biopsy showed diffuse expansion of mesangial matrix by pale, acellular PAS-negative amorphous material, which was congophilic on Congo red stain and gave apple green birefringence on polarization, confirming amyloidosis. Direct immunofluorescence showed nonspecific smudgy deposits of immunoglobulins in the mesangium. There was no light chain restriction observed in the amyloid material. The amyloid material showed strong expression for serum amyloid-associated protein on immunohistochemistry [Figure 2]. Next-generation sequencing showed a compound heterozygous pathogenic variant in the COL7A1 gene with autosomal recessive inheritance. A novel pathogenic heterozygous 5’ splice site variation was detected in intron 103 of the COL7A1 gene, affecting the invariant GT donor splice site of exon 103 (c.7758+2C>G). Another novel likely pathogenic heterozygous missense variation was detected in exon 109 of the COL7A1 gene (chr3:g.48604622C>A), resulting in the amino acid substitution of valine for glycine at codon 2683 (p.Gly2683Val). The in silico predictions of the variant were damage by PolyPhen-2 (HumDiv), scale-invariant feature transform (SIFT), and MutationTaster2. Both the parents were carriers for one of the variants. The diagnosis of secondary renal amyloidosis in a child with RDEB was made. He was managed with prednisolone at 60 mg/day and losartan at 50 mg/day.
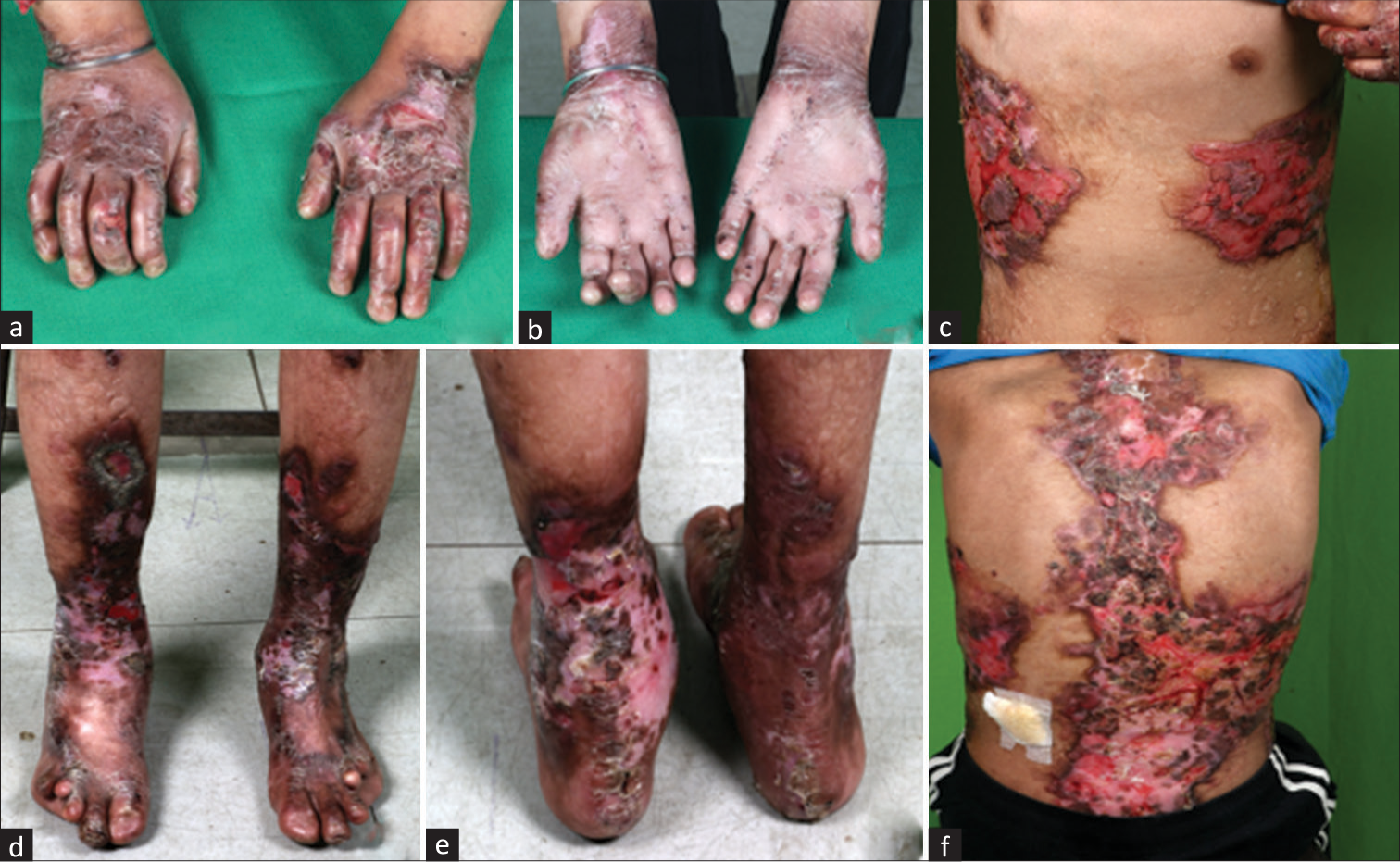
- (a–f) Patient with severe generalized recessive dystrophic epidermolysis bullosa. (a and b) Erosions and scarring of the skin of palms and dorsa of both the hands with contracture of hands, pseudoainhum of distal phalanges, and anonychia. (c and f) Extensive areas of erosions and figurate scarring over the trunk. Note the discrete atrophic albopapuloid macules and papules over the back with a wrinkled surface. (d and e) Nonhealing ulcers over the legs with areas of depigmentation and scarring accompanying nail loss and deformity of toes.
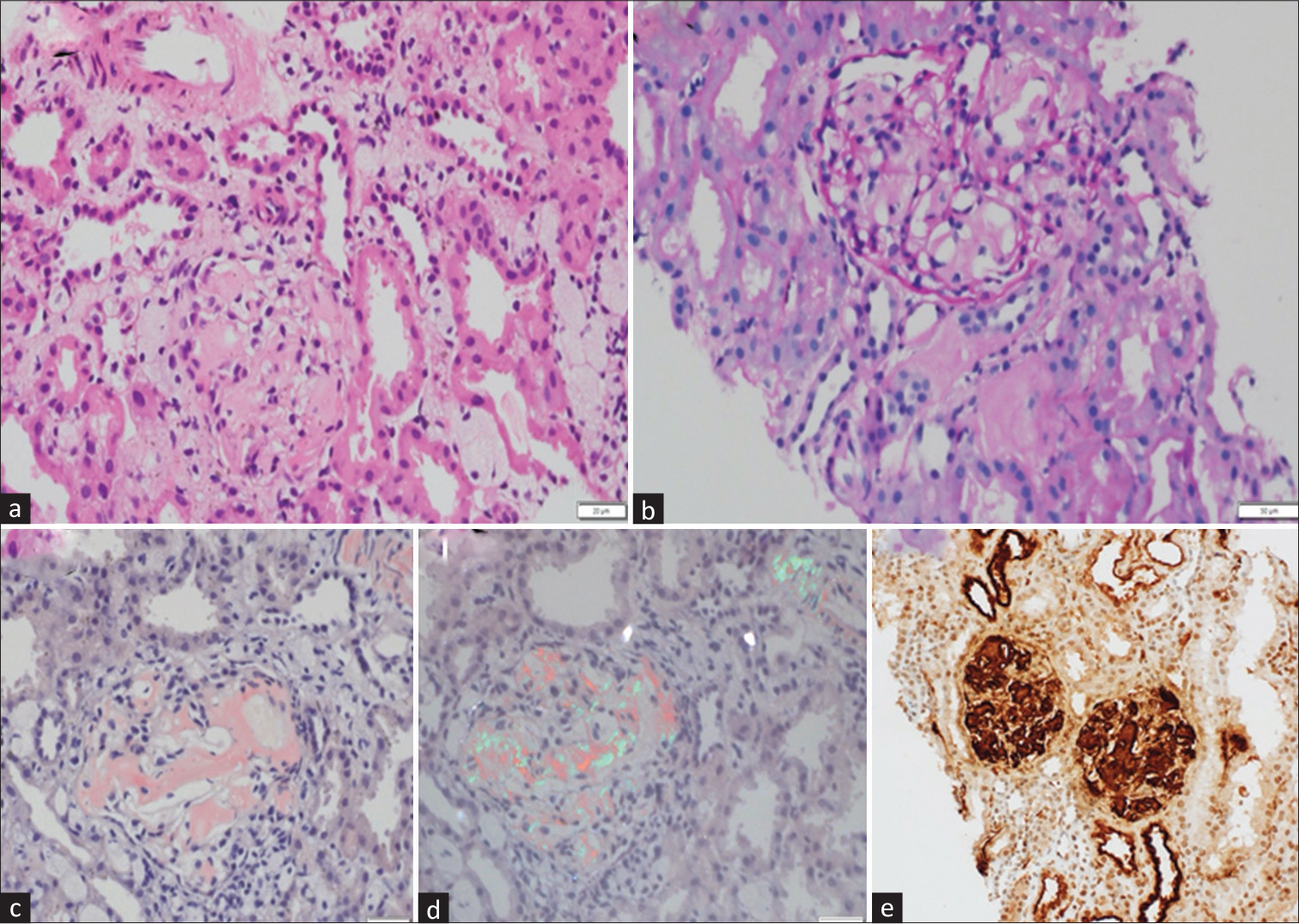
- Renal biopsy. Photomicrographs show (a) acellular amorphous material in the mesangium and basement membranes of the glomerulus, (b) which is PAS negative. Similar material is seen in the interstitium also. (c) This material was congophilic and showed (d) apple green refringence on polarized microscopy, thereby confirming it to be amyloid. (e) SAA immunostaining showed strong reactivity in the amyloid material (H and E - a, PAS - b, Congo red - c and d, IHC-SAA - e. Original magnification 200X). H and E = hematoxylin and eosin, PAS = Periodic Acid Schiff, IHC = immunohistochemistry, SAA = serum amyloid A.
Discussion
Our index case had a compound heterozygous mutation detected in the COL7A1 gene suggestive of RDEB complicated with secondary renal amyloidosis. The incidence of RDEB is 3.05 per 1 million live births.8 One of the rare but important complications of EB is secondary amyloidosis. Renal amyloidosis is caused due to the deposition of AA amyloid, and it is usually a life-threatening condition in patients with RDEB due to the rapid progression of the underlying renal disease as well as secondary systemic infection.9,10 Renal involvement in the form of proteinuria and hematuria is a known complication of RDEB.11 It has been postulated that the renal pathology is either due to secondary renal amyloidosis or mesangioproliferative glomerulonephritis with IgA deposits. The renal histopathology in the index case was suggestive of renal amyloidosis, which has also been reported in RDEB with systemic amyloidosis.5-7 The elevated level of SAA is associated with increased risk of secondary amyloidosis, which was seen in our index case.12 The progression of renal amyloidosis to end-stage renal disease (ESRD) is a known outcome, and the response to therapies like colchicine has been seen in some patients.5,11 Steroids and other immunomodulatory drugs had been tried for secondary amyloidosis; however, there was no response to steroids, and the immunomodulatory drugs could not be started in view of secondary skin infection.13,14 Due to the recurrent skin infections and chronic systemic inflammation, the SAA levels are persistently elevated, which further increases the risk of secondary amyloidosis.15 In severe cases, dysphagia secondary to esophageal atresia with resultant malnutrition and vision impairment secondary to corneal erosions is seen and was present in our index case too.
The early onset of renal involvement in RDEB with rapid progression should alert the treating physician to screen for renal involvement in children with RDEB for early detection of AA amyloidosis and preventing further progression of the renal disease. Due to the defect in the skin integrity, there is a high risk of secondary skin infection. The complications associated with the disease per se should be tackled in addition to treating the primary disease to improve the quality of life and decrease the morbidity. There has been no definitive treatment for EB, but supportive care should be provided to prevent the complications.
Conclusion
In conclusion, secondary renal amyloidosis does occur in patients with RDEB. For early diagnosis of renal amyloidosis in patients with RDEB, periodic screening with urinalysis should be done.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
References
- Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-27.
- [CrossRef] [PubMed] [Google Scholar]
- Premature termination codons on both alleles of the type VII collagen gene (COL7A1) in three brothers with recessive dystrophic epidermolysis bullosa. J Clin Invest. 1995;95:1328-34.
- [CrossRef] [PubMed] [Google Scholar]
- The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol. 2002;146:267-74.
- [CrossRef] [PubMed] [Google Scholar]
- From clinical phenotype to genotypic modelling: Incidence and prevalence of recessive dystrophic epidermolysis bullosa (RDEB) Clin Cosmet Investig Dermatol. 2019;12:933-42.
- [CrossRef] [PubMed] [Google Scholar]
- Renal amyloidosis in recessive dystrophic epidermolysis bullosa. Dermatology (Basel). 2000;200:209-12.
- [CrossRef] [PubMed] [Google Scholar]
- Recessive dystrophic epidermolysis bullosa complicated with nephrotic syndrome due to secondary amyloidosis. Int J Dermatol. 2000;39:151-3.
- [CrossRef] [PubMed] [Google Scholar]
- Renal amyloidosis complicating a recessive epidermolysis bullosa in childhood. Helv Paediatr Acta. 1983;38:167-70.
- [Google Scholar]
- Inherited epidermolysis bullosa and the risk of death from renal disease: Experience of the National Epidermolysis Bullosa Registry. Am J Kidney Dis. 2004;44:651-60.
- [CrossRef] [PubMed] [Google Scholar]
- Fatal systemic amyloidosis (AA type) in two sisters with dystrophic epidermolysis bullosa. J Am Acad Dermatol. 1995;33:370-2.
- [CrossRef] [PubMed] [Google Scholar]
- The spectrum of renal involvement in epidermolysis bullosa dystrophica hereditaria: Report of two cases. Am J Kidney Dis. 1988;11:437-41.
- [CrossRef] [PubMed] [Google Scholar]
- Epidermolysis bullosa complicated with nephrotic syndrome due to AA amyloidosis: A case report and brief review of literature. Saudi J Kidney Dis Transpl. 2019;30:1450-6.
- [CrossRef] [PubMed] [Google Scholar]
- Serum amyloid-A protein concentration in inflammatory diseases and its relationship to the incidence of reactive systemic amyloidosis. Lancet. 1982;2:231-4.
- [CrossRef] [PubMed] [Google Scholar]
- Colchicine therapy in amyloid nephropathy due to recessive dystrophic epidermolysis bullosa. Pediatr Nephrol. 2003;18:1311-2.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular therapeutics in development for epidermolysis Bullosa: Update 2020. Mol Diagn Ther. 2020;24:299-309.
- [CrossRef] [PubMed] [Google Scholar]
- Nephro-urological complications of epidermolysis bullosa in paediatric patients. Br J Dermatol. 2007;156:143-7.
- [CrossRef] [PubMed] [Google Scholar]




