

Translate this page into:
Renal Complications of Hematopoietic Stem Cell Transplantation: Report of a Case and Review of the Literature
Address for correspondence: Dr. K. K. Venkat, Division of Nephrology and Hypertension, Henry Ford Hospital, CFP-5, 2799 West Grand Boulevard, Detroit, MI 48202, USA. E-mail: kvenkat1@hfhs.org
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
We report the development of minimal change disease superimposed on preexisting chronic kidney disease secondary to chronic calcineurin inhibitor nephrotoxicity in a hematopoietic stem cell transplantation (HSCT) recipient and review the renal complications of HSCT.
Keywords
Chronic calcineurin inhibitor nephrotoxicity
graft-versus-host disease
hematopoietic stem cell transplantation
minimal change disease
nephrotic syndrome

Introduction
Hematopoietic stem cell transplantation (HSCT), autologous or allogeneic, is well established as a therapy for a variety of malignant and benign disorders.[1] Graft-versus-host disease (GVHD), wherein donor-origin immune-competent cells target “foreign” human leukocyte antigens in recipient tissues (mainly skin, gastrointestinal tract, and liver), is the major problem in allogeneic HSCT, in contrast to rejection of the transplanted organ by the recipient's immune system in solid organ transplantation.[1] We report a HSCT recipient who developed nephrotic syndrome and acute kidney injury (AKI) due to minimal change disease (MCD) superimposed on chronic kidney disease (CKD) secondary to chronic calcineurin inhibitor (CNI) nephrotoxicity in the setting of chronic GVHD and review the renal complications of HSCT.
Case Report
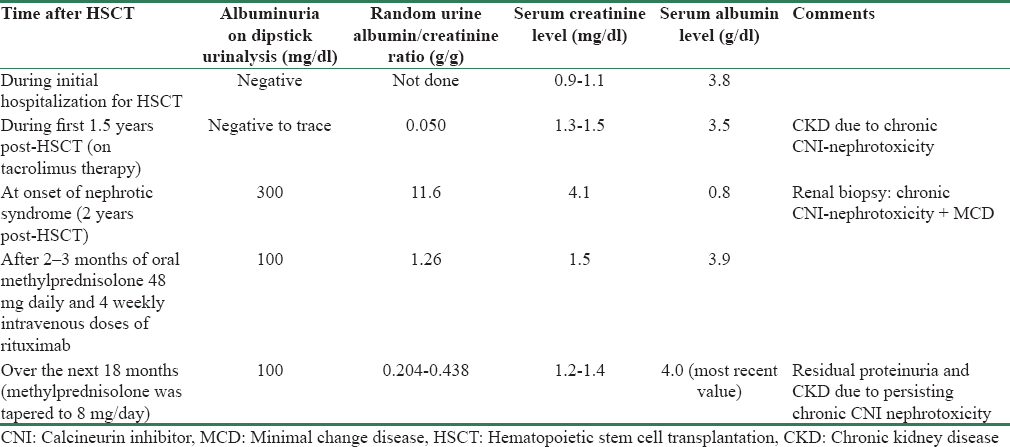
A 40-year-old-man received an allogeneic, unrelated donor HSCT for thalassemia intermedia because transfusion dependence and hemosiderosis persisted after splenectomy. HSCT preconditioning with fludarabine, busulfan, and rabbit anti-thymocyte globulin (no radiation) was followed by tacrolimus and methotrexate prophylaxis for 1 month. Thalassemia-associated anemia and microcytosis normalized post-HSCT. GVHD (predominantly cutaneous with mild liver involvement) developed 1-month post-HSCT. Over the next 2 years, GVHD was treated with daily oral tacrolimus, methylprednisolone, and sirolimus (latter discontinued 22 months post-HSCT; at the time of sirolimus discontinuation, our patient had no edema and the most recent available urinalysis had shown only trace albuminuria) and subcutaneous etanercept and extracorporeal photopheresis intermittently. Other medications included acyclovir, amlodipine, and alendronate. Serum creatinine (SCr) level during the initial hospitalization for HSCT ranged between 0.9 and 1.1 mg/dl. Over the next 1.5 years, SCr fluctuated between 1.3 and 1.5 mg/dl, and occasionally up to 2.0 mg/dl, with multiple urinalyses showing negative to trace albuminuria and an unremarkable sediment.
Two years post-HSCT, lower extremity swelling and 18.2 kg weight gain developed over 3 weeks. Examination revealed severe, symmetrical edema up to the groins, cutaneous features of GVHD, and blood pressure of 132/84 mm Hg. SCr had increased to 4.1 mg/dl from 1.4 mg/dl a month earlier. Urinalysis showed 300 mg/dl albuminuria, 4–6 red blood cells/high power field, and 4–5 granular and 0–3 hyaline casts/low power field. Random urine albumin/creatinine ratio (UAlb/Cr) was 11.6 g/g (previously 50 mg/g). Serum albumin (SAlb) level was 0.8 g/dl (3.5 g/dl 6 weeks earlier). Other relevant results were markedly elevated serum cholesterol and triglyceride levels markedly elevated; tacrolimus trough level 7.7 ng/ml; anti-nuclear, anti-neutrophil cytoplasmic, HIV and hepatitis-C antibodies, and hepatitis-B surface antigen negative; serum C3 and C4 complement levels normal; serum and urine negative for monoclonal proteins and light chains. Renal ultrasonography revealed normal renal lengths bilaterally without hydronephrosis. Because of elevated SCr, tacrolimus, alendronate, and acyclovir were stopped. Lisinopril, furosemide, and atorvastatin were added.
Renal biopsy
Light microscopic examination revealed nodular hyaline arteriolosclerosis [Figure 1a and b] and “striped” interstitial fibrosis and tubular atrophy (IF-TA) with 9/28 globally sclerotic glomeruli [Figure 1c]. Direct immunofluorescence showed scattered, predominantly mesangial, nonspecific staining for IgG, IgA, IgM, c1q, C3 and C4 complements, and fibrinogen. Electron microscopy revealed widespread foot process effacement with no electron-dense deposits [Figure 1d]. The final renal biopsy diagnoses were nodular hyaline arteriolosclerosis, global glomerulosclerosis and band-like IF-TA, compatible with chronic CNI-nephrotoxicity, and widespread foot process effacement compatible with MCD.

- Photomicrographs from the patient's kidney biopsy. (a) Nodule of hyaline arteriolosclerosis at the vascular pole of the glomerulus (arrow). (b) Nodular hyaline arteriolosclerosis (arrows) - higher power view. (c) Band-like (striped) tubular atrophy and interstitial fibrosis along the top and bottom edges of the biopsy core (black arrows) and a globally sclerosed glomerulus (white arrow). (d) Diffuse podocyte (foot process) effacement/fusion on electron microscopy (arrows). There are no electron-dense deposits
Following biopsy, oral methylprednisolone was increased to 48 mg daily (tapered over 12 weeks to 10 mg daily) and rituximab was given intravenously (375 mg/m2, 4 weekly doses). Within 3 months, edema resolved completely (permitting furosemide discontinuation), SAlb normalized (3.9 g/dl), and UAlb/Cr decreased to 1.26 g/g. Over the next 18 months, UAlb/Cr has fluctuated between 204 and 438 mg/g (without lisinopril) with most recent SAlb of 4.0 g/dl. SCr decreased to 1.5 mg/dl 8 weeks postrituximab and has fluctuated between 1.2 and 1.4 mg/dl over the next 18 months. Cutaneous features of GVHD also improved and are currently well controlled on methylprednisolone 8 mg/day alone. Blood pressure is normal without medications. Hematological test results which became normal approximately 2 months post-HSCT have remained normal till now, indicating a well-functioning HSCT (based on peripheral blood cell counts only; follow-up bone marrow/flow cytometry/chimerization studies not done).
Table 1 shows selected laboratory values in our patient at different time intervals after HSCT.
Discussion
HSCT may be complicated by a variety of renal disorders.[23456] AKI of different etiologies is common in the first 100 days following HSCT (incidence of 21%–73%). Thereafter, CKD becomes increasingly prevalent. Glomerular disease and nephrotic syndrome are uncommon complications of HSCT. Thus, our patient developed both a common (CKD) and an uncommon (MCD) renal complication of HSCT.
Pathologically, chronic CNI-nephrotoxicity is characterized by nodular preglomerular arteriolar hyalinosis originating in the media, global, and/or focal segmental glomerulosclerosis (FSGS) and IF-TA (initially band-like/“striped,” eventually becoming diffuse).[7] In transplanted kidneys, these changes are nonspecific, occurring also in chronic rejection, older donor kidneys, polyoma virus nephropathy, and transplant renal artery stenosis.[8] Diabetes and hypertension also cause arteriolar hyalinosis (originating in the subendothelium). However, nodular arteriolar hyalinosis affecting the native kidneys (which are not subject to rejection) of patients on long-term CNI-therapy (such as ours) favors the diagnosis of chronic CNI-nephrotoxicity.[9]
The diagnosis of MCD is difficult when it is superimposed on preexisting pathological changes as in our patient. The term podocytopathy has been used instead of MCD when an MCD-like disorder is superimposed on a different renal disorder such as lupus nephritis.[10] The diagnosis of superimposed MCD/podocytopathy in this patient is supported by the following features. While nephrotic syndrome develops insidiously in most glomerulopathies, sudden onset of severe nephrotic syndrome is typical of MCD.[11] Widespread podocyte effacement may be seen in any heavily proteinuric disorder.[12] However, as an isolated finding without pathological features of other glomerulopathies, it defines MCD. Rapid resolution of proteinuria following increased corticosteroid dose and rituximab also suggests MCD, which is corticosteroid-responsive in most patients and also responds to rituximab.[13] Rituximab was chosen in our patient because it is effective in treating both MCD and GVHD. These features support the diagnosis of MCD in our patient. Nephrotic syndrome was associated with reversible AKI in our patient, a complication most commonly reported in MCD (especially in adults).[1114] AKI in this setting was previously attributed to anasarca-associated renal interstitial edema but is now believed to be due to ischemic tubular injury.[1114] MCD typically resolves without residual proteinuria or renal dysfunction. Persistence of CKD and minimal albuminuria in our patient was due to chronic CNI-nephrotoxicity, which may not be reversible even after discontinuation of CNI. Glomeruli in MCD usually appear unremarkable on light microscopy. The 32% global glomerulosclerosis in our patient's biopsy was also due to preexisting chronic CNI-nephrotoxicity.
In addition to generic causes of AKI, hepatic sinusoidal obstruction syndrome which mimics hepatorenal syndrome (putative mechanisms: Sinusoidal endothelial injury due to drugs or radiation used for preconditioning and/or acute GVHD), tumor-lysis syndrome caused by preconditioning chemotherapy for leukemia or lymphoma, acute thrombotic microangiopathy (TMA) caused by CNI and/or acute GVHD-associated generalized endothelial injury, and acute GVHD-mediated direct renal tubular injury are special etiological considerations for AKI occurring in the initial 100 days post-HSCT.[26] CKD develops after the first 3 months in 7 to 48% of HSCT recipients and progresses to end-stage renal disease in approximately 4% of patients with CKD.[346] The major causes of post-HSCT CKD are chronic CNI-nephrotoxicity (as in our patient), chronic TMA (“bone marrow transplant nephropathy”) attributed to chronic endothelial injury due to CNI and/or chronic GVHD, and (uncommonly) BK-polyoma virus nephropathy.[346]
Nephrotic syndrome is rare after HSCT, occurring only in 0.4%–6.0% of adult HSCT-recipients.[6] It can develop within a few months of HSCT, but usually occurs after the 1st year, almost exclusively in the setting of chronic GVHD, and has an incidence of 0.4%–6% with biopsy revealing membranous nephropathy in approximately two-thirds and MCD in one-quarter of patients.[56] A number of observations suggest that common immunopathogenic mechanisms may underlie both GVHD and glomerulopathies complicating HSCT:[615] (1) Nephrotic syndrome develops only rarely in the absence of coexisting GVHD. (2) Increased production of potentially glomerulopathic cytokines such as interferon and tumor necrosis factor has been documented in GVHD. (3) Renal tubulointerstitial lymphoplasmacytic infiltrate of donor origin has been demonstrated in the biopsy of some patients with HSCT-associated nephrotic syndrome. (4) Reduction/discontinuation of immunosuppression has been reported to precede the development of nephrotic syndrome in association with exacerbation of GVHD. (5) Finally, reinstitution of immunosuppression frequently results in remission of both the nephrotic syndrome and GVHD (as in our patient). We did not perform any studies aimed at identifying the immunopathogenesis of MCD/podocytopathy (such as CD4/CD8 cell subtyping in the peripheral blood or CD80 staining of the renal biopsy) in our patient. There are rare reports of FSGS, mesangial proliferative, or crescentic glomerulonephritis in association with chronic GVHD, but a cause-effect relationship between GVHD and these glomerulopathies is not established.[56] Sirolimus-associated nephrotic syndrome is unlikely in our patient since this drug was discontinued a few months before nephrotic syndrome developed. Etanercept and photopheresis used in this patient for treating GVHD have not been associated with the development of nephrotic syndrome. Given a variety of renal disorders that may cause nephrotic syndrome in HSCT recipients and since it is impossible to diagnose these disorders on clinical grounds, nephrology consultation and renal biopsy are appropriate when this complication develops. The clinical presentation, prognosis, and response to therapy of HSCT-associated MCD appear to be similar to idiopathic MCD, with reinstitution/intensification of Immunosuppression causing prompt remission of nephrotic syndrome.[5] However, persistence of AKI, CKD, and proteinuria, irrespective of etiology, adversely affects long-term survival of HSCT patients.[6]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Acute kidney injury in hematopoietic cell transplantation. Semin Nephrol. 2010;30:615-26.
- [Google Scholar]
- Chronic kidney disease after hematopoietic stem cell transplantation. Semin Nephrol. 2010;30:627-34.
- [Google Scholar]
- The kidney effects of hematopoietic stem cell transplantation. Adv Chronic Kidney Dis. 2014;21:96-105.
- [Google Scholar]
- Nephrotic syndrome associated with chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2006;38:351-7.
- [Google Scholar]
- Renal complications of hematopoietic-cell transplantation. N Engl J Med. 2016;374:2256-67.
- [Google Scholar]
- Primer: Histopathology of calcineurin-inhibitor toxicity in renal allografts. Nat Clin Pract Nephrol. 2006;2:398-404.
- [Google Scholar]
- Specificity of histological markers of long-term CNI nephrotoxicity in kidney-transplant recipients under low-dose cyclosporine therapy. Am J Transplant. 2011;11:2635-46.
- [Google Scholar]
- Tacrolimus and cyclosporine nephrotoxicity in native kidneys of pancreas transplant recipients. Clin J Am Soc Nephrol. 2011;6:101-6.
- [Google Scholar]
- Clinical-morphological features and outcomes of lupus podocytopathy. Clin J Am Soc Nephrol. 2016;11:585-92.
- [Google Scholar]
- Adult minimal-change disease: Clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol. 2007;2:445-53.
- [Google Scholar]
- Podocyte foot process effacement as a diagnostic tool in focal segmental glomerulosclerosis. Kidney Int. 2008;74:1568-76.
- [Google Scholar]
- Rituximab for minimal change disease in adults: Long-term follow-up. Nephrol Dial Transplant. 2014;29:851-6.
- [Google Scholar]
- Minimal-change disease in adolescents and adults: Epidemiology and therapeutic response. Clin Kidney J. 2013;6:469-72.
- [Google Scholar]
- Nephrotic syndrome after hematopoietic cell transplantation: Do glomerular lesions represent renal graft-versus-host disease? Clin J Am Soc Nephrol. 2006;1:685-94.
- [Google Scholar]




