

Translate this page into:
Renal disease masquerading as pyrexia of unknown origin
Address for correspondence: Dr. Deepa Korivi, New Wing, Bombay Hospital, New Marine Lines, Mumbai - 400 020, Maharashtra, India. E-mail: udeepa21@rediffmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Pyrexia of unknown origin is a challenging clinical problem. Infections, malignancies, and connective tissue diseases form the major etiologies for this condition. We report a case of a 57-year-old diabetic male who presented with fever of unknown origin for several months. The course of investigations led to a kidney biopsy which clinched the cause of his fever as well as the underlying diagnosis. The light microscopy findings of expansile storiform fibrosis with a dense inflammatory infiltrate suggested the diagnosis which was confirmed by positive staining of Immunoglobulin G4, the dense lympho-plasmacytic infiltrate and elevated serum IgG4 concentrations. A course of steroids followed by mycophenolate mofetil as maintenance immunosuppression rendered the patient afebrile with improvement of renal function.
Keywords
Autoimmune pancreatitis
corticosteroids
immunoglobulin G4 related disease
interstitial fibrosis
tubulointerstitial nephritis

Introduction
Immunoglobulin G4 -related disease (IgG4RD) is a novel disease entity associated with high serum IgG4 levels. It can present in a variety of clinical, radiologic, and biochemical fashions. The kidney is a distinct target organ affected by this disease entity and we report one such patient with an unusual presentation.
Case Report
A 57-year-old gentleman was referred with high grade intermittent fever with chills since 3 months. He was a longstanding type 2 diabetic, on oral hypoglycemic agents with a fairly good sugar control. He did not have any constitutional symptoms like weight loss or anorexia. He was normotensive and had no specific urinary symptoms. Other than the fever for which he consumed paracetamol, he was fairly asymptomatic. There were no clinical clues to the etiology for the fever on systemic examination.
The complete blood count of the patient showed an Hemoglobin 11 g/dL, total leukocyte count 10,100 (polymorphs 85, lymphocytes 13% and eosinophils 2%) and a normal platelet count of 2.73 lacs. Liver function tests were essentially normal with a bilirubin of 0.7 mg/dL, alanine aminotransferase/ALT of 36 and 31 IU/L, serum albumin 3.84 g/dL, and serum globulin 2.46 g/dL. Renal function tests showed BUN 55 mg/dL, serum creatinine 1.5 mg/dL, and normal serum electrolytes. The urine analysis was essentially bland with no albumin, 1-2 pus cells, and no red cells.
Test results during these 3 months revealed an erythrocyte sedimentation rate of 90 mm at the end of 1 h and two high C-reactive protein levels of 35.8, 24, and 18.5 mg/L (Normal up to 6 mg/L). Complete blood count, urine examination, chest radiology, and 2D Echo performed to investigate the cause of the fever were all normal. Blood and urine cultures were also negative. Serum Ferritin and complement levels were normal. anti-nuclear antibodies and anti-neutrophil cytoplasmic antibody were negative. angiotensin converting enzyme levels were normal. Tumor marker profile was negative. Enzyme immunoassays for tuberculosis and brucellosis were negative. He was treated empirically with antibiotics during this period with no permanent relief.
The abdominal ultrasound was also essentially normal. A whole body computed tomography (CT) scan revealed multiple lymph nodes measuring 1.5-4 cm in the mediastinum and retrocarinal region reported to be of inflammatory/infective etiology. Fine needle aspiration of the lymph node was reported as non-specific lymphadenitis. The contrast CT abdomen revealed heterogeneous enhancement of both kidneys with hypodensities at the lower pole [Figure 1].
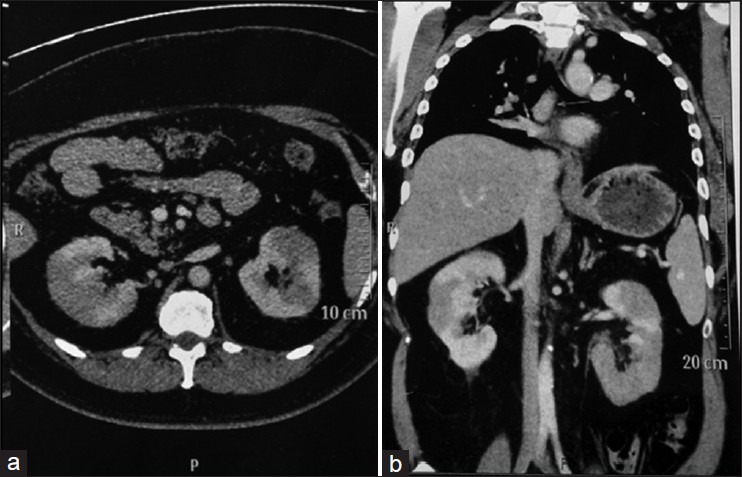
- Contrast computed tomography abdomen showing heterogeous enhancement of the kidneys with hypodensities at the lower pole
Given the clinical presentation and the clinical possibilities of vasculitis and lymphoma, and the kidney findings on CT, a renal biopsy was performed. The conspicuous feature was presence of expansile interstitial fibrosis with a plasma cell rich interstitial infiltrate that were accompanied by a few lymphocytes and occasional eosinophil [Figure 2a]. Some areas of the biopsy showed storiform fibrosis. There were neither neutrophils nor granulomas. The fibro inflammatory process was seen to extend into the peri-nephric tissue and had resulted in significant tubular loss. The remaining tubules showed thickening of their basement membranes. The glomeruli revealed diffuse capillary wall thickening but there were no changes of diffuse or nodular diabetic glomerulopathy. The arteries revealed mild medial thickening. The pathologic features raised the suspicion for IgG4 related tubulointerstitial nephritis. Immunofluorescence was negative for IgG, IgA, IgM, C3 and C4. IgG4 immunohistochemistry was performed which showed numerous IgG4 expressing plasma cells amounting to at least 15 per high power field (HPF) [Figure 2b]. The pathologic diagnosis was IgG4 related tubulointerstitial nephritis. Corresponding serum IgG4 level was 5.9 g/L (N-0.01-1.3 g/L). The diagnosis of IgG4 mediated nephropathy was made on the basis of the clinical history, CT scan findings, histopathology, immunohistochemistry, and serology. He was initiated on a dose of 40 mg prednisolone (0.5 mg/kg/day) and his fever defervesced within 24 h. The steroids were continued and tapered over the next 5 months. His serum creatinine touched a baseline of 1.2 mg/dL at the end of 2 months therapy with steroids. Although an attempt was being made to taper down the steroids further, his fever recurred at a steroid dose of 5 mg/day. He was therefore initiated on Mycophenolate Mofetil (MMF) with a dose of 1 g b.i.d to which his fever responded again. Steroids were stopped by the end of the 5th month and MMF was continued. The serum IgG4 levels progressively declined from a peak of 5.9 g/L to 2.04 followed by 1.55 g/L by the end of the 4th month. The patient continued to be on a dose of 2 g of MMF per day. The CT scan repeated after 6 months showed a significant regression in the mediastinal and retrocarinal lymphadenopathy and resolution of the original hypodense foci in the kidneys.
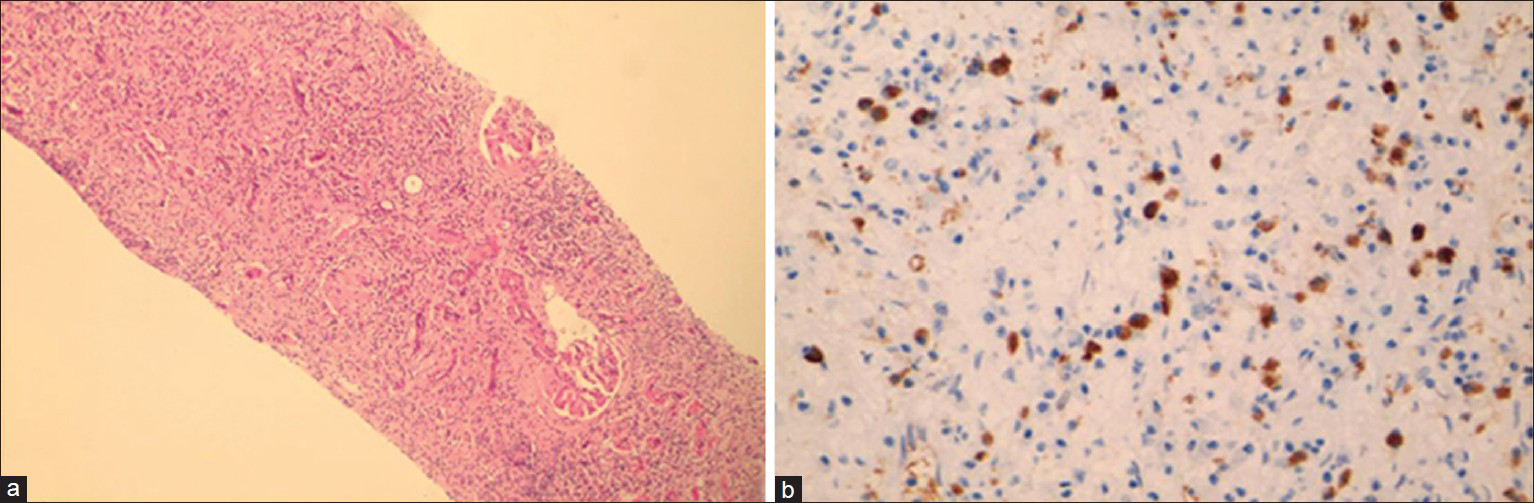
- (a) Expansile interstitial fibrosis (H and E, ×100), (b) Plasma cells with expression for immunoglobulin G4 (×400s)
Discussion
IgG4RD is an increasingly recognized syndrome of unknown etiology comprised a collection of disorders that share specific clinical, serologic, and pathologic features. The commonly shared features include tumor like swelling of the involved organs, a lymphoplasmacytic infiltrate rich in IgG4 positive plasma cells, and variable degrees of fibrosis.[1] Glucocorticoid responsiveness has been considered as one of the diagnostic criterion for this disorder.
Renal involvement in IgG4RD is relatively uncommon when compared with autoimmune pancreatitis which is the most studied group with the latter being linked to elevated serum IgG4 concentrations as early as 2001. The myriad of renal presentations include hydronephrosis secondary to retroperitoneal fibrosis that may represent extra-renal extension of the fibro inflammatory pathology and tubulointerstitial nephritis.[2] Rarely, patients show multiple tumor like low density areas on imaging like our patient. The renal biopsy in such cases reveals aggregates of lymphocytes and plasma cells in the renal interstitium. Asymptomatic lymphadenopathy is common, occurring in 80% of patients and may be the only initial manifestation. Mediastinal, hilar, intra-abdominal, and axillary lymphadenopathy are the common sites and are readily detectable on Gallium 67 scanning.[3] In fact, the combination of renal involvement and lymphadenopathy should arouse clinical suspicion of IgG4-related disease. Interestingly, the renal imaging findings may suggest possibility of vasculitis.
The overall disease epidemiology remains largely undefined. It is more often described as occurring in middle aged and older men. The majority of patients with IgG4-RD have elevated serum IgG4 concentrations, but the range varies widely. Approximately 30% of patients have normal serum IgG4 concentrations, despite classic histopathologic and immunohistochemical findings.[145] IgG4 has been postulated to have a role in tolerance to allergens. However, as a corollary it has been found that up to 40% of patients with IgG4RD have peripheral eosinophilia. Immune complex deposits in the pancreas, kidneys, and certain other affected tissues have been reported. The number of IgG4-positive plasma cells per HPF regarded as sufficient for the diagnosis of IgG4RD varies somewhat from tissue to tissue; generally, the minimum for making the diagnosis for most tissues is from 30 to 50 IgG4-positive cells/HPF. However, only 10 IgG4 positive plasma cells/HPF may be sufficient in some organs or tissues, including the kidney.[6] There are increased numbers of T regulatory cells (Tregs) in peripheral blood and increased levels of cytokines produced by Tregs, including interleukin (IL)-10 and transforming growth factor-β in affected tissues. The Th 2 cytokines, Tregs, and IL-10 help support IgG4 production.[17]
Corticosteroids form the mainstay treatment and most patients respond within several weeks. Although IgG4 concentrations become lower with glucocorticoid treatment in the great majority of patients in whom they are elevated at baseline, they remain above normal values in most patients. A multicenter study from Japan showed that IgG4 levels failed to normalize in 115 of 182 patients (63%) treated with glucocorticoids.[8] Azathioprine and MMF have been used for patients who require chronic therapy to avoid the adverse effects of steroids. B cell depletion therapy with rituximab is an effective treatment for patients refractory to glucocorticoids and standard immunosuppression.[9]
The natural history of IgG4RD has not been well defined. Some patients improve spontaneously without treatment but many relapse in time. Several types of lymphomas especially non-Hodgkin's variety have been reported in these patients 3-5 years after the diagnosis of IgG4RD. It would therefore benefit if these patients are on a regular follow-up.[10]
Pyrexia of unknown origin (PUO) is a diagnostic challenge. In the general population infections form the major etiology followed by autoimmune diseases and malignancies. In elderly subjects however, unlike in the young, a precise diagnosis can be made 87-95% of the times. In a study carried out in elderly patients, infections constituted 25-35%, connective tissue disorders constituted 25-35%, and malignancies accounted for 12-23% of the cases.[11] As many of these diseases are treatable it is recommended that etiology of PUO in elderly should be investigated further. In this patient, PUO was the presenting manifestation and the CT finding of wedge shaped hypodensities within the kidney, steered the investigative algorithm towards the kidney as the organ of interest. As renal infections, vasculitis, and septic emboli could present in this fashion with fever and renal hypodensities, the kidney biopsy showed a clinically unsuspected diagnosis for the presenting symptoms. Fever in IgG4RD usually presents subacutely and patients are not constitutionally ill. It is an unusual early presentation.[1] The disease is often identified incidentally through radiologic findings or unexpectedly in pathologic specimens. This patient had an uncommon presentation of an even more uncommon disease.
Conclusion
IgG4RD is a newly recognized fibro inflammatory condition characterized by tumefactive lesions, a dense lymphoplasmacytic infiltrate, and storiform fibrosis of the tissue. It is an unrecognized condition about which knowledge is increasing. High index of suspicion is required especially with rare extra pancreatic manifestations of this disease. Timely diagnosis, treatment, and disease monitoring will play a significant role in influencing the long-term outcome of the patient.
Acknowledgments
The authors wish to acknowledge histopathologists Vinita Pant and Girish Mujumdar for their assistance in the specialized staining and interpretation of the biopsy specimen.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78:1016-23.
- [Google Scholar]
- Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:671-81.
- [Google Scholar]
- Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol. 2011;23:108-13.
- [Google Scholar]
- High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732-8.
- [Google Scholar]
- A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:57-66.
- [Google Scholar]
- Circulating naïve and CD4+CD25 high regulatory T cells in patients with autoimmune pancreatitis. Pancreas. 2008;36:133-40.
- [Google Scholar]
- Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755-62.
- [Google Scholar]
- Possible association between IgG4-associated systemic disease with or without autoimmune pancreatitis and non-Hodgkin lymphoma. Pancreas. 2009;38:523-6.
- [Google Scholar]




