

Translate this page into:
Secondary Membranous Nephropathy and Immunodeficiency due to a Novel Biallelic Variant in CARMIL2
Corresponding author: Vivekananda Bhat, Department of Medical Genetics, Kasturba Medical College, Manipal, Manipal Academy of Higher Education, Manipal, Karnataka, India. E-mail: bhat.vivekananda@manipal.edu
-
Received: ,
Accepted: ,
How to cite this article: Rao LP, Kothiwale V, Radhakrishnan P, Rangaswamy D, Shukla A, Bhat V. Secondary Membranous Nephropathy and Immunodeficiency due to a Novel Biallelic Variant in CARMIL2. Indian J Nephrol. 2024;34:667-9. doi: 10.25259/ijn_542_23
Abstract
The cytosolic capping protein, Arp2/3 and myosin-I linker protein 2 or CARMIL2 plays an important role in T/B/NK cell function. Biallelic disease causing variants in CARMIL2 are known to cause immunodeficiency 58. We report a 13-year-old girl with recurrent infections, dermatitis and nephrotic syndrome since childhood. Her renal biopsy was suggestive of membranous nephropathy. Exome sequencing showed a homozygous novel stopgain variant, c.520C>T in CARMIL2 (NM_001013838.3). We expand the phenotypic spectrum of CARMIL2 related immunodeficiency to include membranous nephropathy secondary to probable immune dysregulation.
Keywords
Immunodeficiency 58
Immune dysregulation
Exome sequencing
Autoimmune hemolytic anemia
Nonsense variant

Introduction
The cytosolic capping protein, Arp2/3 and myosin-I linker protein 2 (CARMIL2), also known as RLTPR, is involved in actin polymerization dynamics and thereby regulates cytoskeletal organization, endocytosis, and cell migration. CARMIL2 is required for CD28-mediated co-stimulation of NF-κB signaling in T cells, which is important for naive T cell activation, proliferation, maturation into T memory cells, and differentiation into T helper (Th) and T regulatory cells (Treg). It is also involved in B and NK cell function.1 Biallelic loss of function variants in CARMIL2 causes immunodeficiency 58 (MIM# 618131) characterized by immunodeficiency and immune dysregulation.1,2 Clinical presentations include recurrent bacterial, viral, and fungal infections; eczematous dermatitis; disseminated Epstein–Barr virus-associated smooth muscle tumors; and early-onset inflammatory bowel disease.2
Case Report
We evaluated a 13-year-old girl born to a non-consanguineous couple [Figure 1a] with recurrent episodes of upper respiratory tract infections, extensive seborrheic dermatitis, and eczema, beginning at 9 months of age. At 7 years of age, she developed sudden-onset facial puffiness, lower extremity swelling, and abdominal distention. She was noted to have anasarca, pallor, generalized lymphadenopathy, atopic dermatitis, and oral candidiasis. Her laboratory investigations were consistent with nephrotic syndrome. Light microscopy examination of the renal biopsy specimen revealed diffuse capillary wall thickening and focal segmental mesangial proliferation, consistent with membranous nephropathy. Immunofluorescence showed glomerular tufts indicative of finely granular deposits of IgG, IgM, C3, and traces of C1q along capillary loops. Due to the negative PLA2R staining, coupled with subendothelial deposits, a secondary form of membranous nephropathy was considered for which she was started on oral prednisolone over a period of 6 weeks and tapered after remission. Institutional ethics approval was obtained prior to the study.
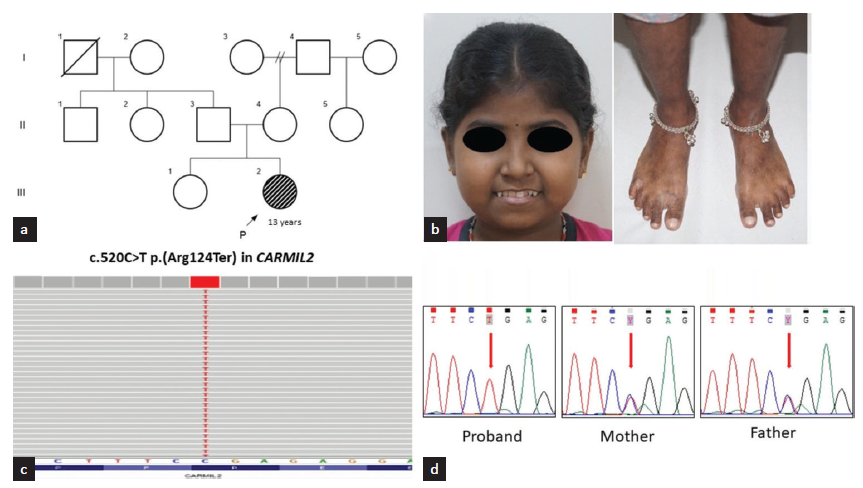
- (a) Pedigree of the family; (b) Clinical photographs of the proband showing facial puffiness, hyperpigmented macules over the lower limbs (c) Integrated genomic viewer image showing sequence variant c.520C>T p.(Arg124Ter) in exon 7 of CARMIL2 in homozygous state; (d) Sanger chromatograms showing the same in homozygous state in the proband and heterozygous state in her parents.
At 13 years of age, she had a relapse of nephrotic syndrome, and steroids were restarted. She had an episode of hypertensive crisis leading to an episode of generalized tonic-clonic seizure. On examination at 13 years, her weight was 24.2 kg (−3.23 SD), height was 122 cm (−5 SD), and head circumference was 53 cm (−0.47 SD). She had short stature, pallor, oral thrush, healed pyoderma rashes, and bilateral cervical lymphadenopathy [Figure 1b]. Her laboratory results showed 24-hour urine protein level of 63 mg/m2/h, hypoalbuminemia (serum albumin: 2.9 g/dL (3.5–5 g/dL); severe anemia (hemoglobin 3.8 g/dL (11–15 g/dL)), elevated erythrocyte sedimentation rate (104 mm/h), and a positive directs Coombs test. Her antinuclear antibody was negative. Peripheral smear showed normocytic normochromic anemia with anisocytosis, red cell clumping, polychromasia, and spherocytosis. Immunological workup was suggestive of altered T-cell subsets and NK cells [Table 1]. Electroencephalogram was indicative of focal epilepsy. Magnetic resonance imaging of the brain showed cerebral atrophy and periventricular subcortical hyperintensities. Oral prednisolone was restarted along with antihypertensives and antiepileptics.
| Test name | Observed value | Reference range |
|---|---|---|
| IgA | <1 mg/dL ↓ | 58–358 mg/dl |
| IgG | 524 mg/dL ↓ | 759–1550 mg/dL |
| C3 | 68 mg/dL ↓ | 90–180 mg/dl |
| CD44 | 3164.74/uL | 2140–3816/uL |
| Absolute CD3 cells count | 1648.7/uL | 1280–2686/uL |
| CD4/CD8 ratio | 0.81 ↓ | 0.9–1.7 |
| B cells | 835.27/uL ↑ | 177–511/uL |
| NK cells | 127.20/uL ↓ | 184–538/uL |
IgA: Immunoglobulin A, IgG: Immunoglobulin G, C3: Complement 3, CD44: cluster of differentiation 44, CD3: cluster of differentiation 3, CD4: Clusters of differentiation 4, CD8: Clusters of differentiation 8, B cells: bursa derived cells, NK cells: Natural killer cells
Singleton exome sequencing showed a novel stop gain variant, c.520C>T p.(Arg124Ter) in exon 7 of CARMIL2 (NM_001013838.3) in the homozygous state in the proband. Parents are heterozygous for this variant [Figure 1c and d]. This variant is not present in the homozygous state in gnomAD or in our in-house database of 3160 exomes. In-silico tools (Mutation taster, CADD) are consistent in predicting the variant to be damaging to CARMIL2 protein function. This variant is predicted to lead to premature termination of the CARMIL2 transcript, which may either trigger nonsense-mediated mRNA decay or result in a truncated protein. The clinical and laboratory findings are in concordance with immunodeficiency 58, caused by biallelic variants in CARMIL2. The variant was classified as ‘likely pathogenic’ by using the standards and guidelines for the interpretation of sequence variants by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.3
Discussion
Biallelic disease-causing variants in CARMIL2 lead to impaired T-cell activation, proliferation, and cytokine response, thereby resulting in reduced development of regulatory and memory T cells.2 The immune-related phenotype is a spectrum ranging from immunodeficiency affecting the T, B, and NK cells to immune dysregulation. Early onset of inflammatory bowel disease arising from immune dysregulation or triggered by infections have also been reported.4
The proband presented with classical signs of immunodeficiency and chronic skin lesions. We report, for the first time, secondary membranous nephropathy as one of the clinical presentations of CARMIL2-related immunodeficiency 58. We hypothesize that a reduced number of Treg cells can lead to the breakdown of immune tolerance leading to the secondary phenotype of membranous nephropathy and autoimmune hemolytic anemia.
Genetic testing has important implications in individuals with suspected immunodeficiency and membranous nephropathy. It provides a definitive diagnosis, suggests available treatment options, provides an understanding of the natural history of the disease, allows focused counseling, and aids in providing the exact risk of recurrence and prenatal testing in future pregnancies of parents.
We expand the phenotypic spectrum of immunodeficiency 58 caused by the biallelic pathogenic variant in CARMIL2 with an additional feature of membranous nephropathy secondary to probable immune dysregulation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
National Institutes of Health, United States, has funded the study, “Genetic Diagnosis of Neurodevelopmental Disorders in India” (1R01HD093570-01A1).
Conflicts of interest
There are no conflicts of interest.
References
- Novel CARMIL2 loss-of-function variants are associated with pediatric inflammatory bowel disease. Sci Rep. 2021;11:5945.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Human CARMIL2 deficiency underlies a broader immunological and clinical phenotype than CD28 deficiency. J Exp Med. 2023;220:e20220275.
- [CrossRef] [PubMed] [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- CARMIL2 deficiency presenting as very early onset iInflammatory bowel disease. Inflamm Bowel Dis. 2019;25:1788-95.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




