

Translate this page into:
Successful Management of Adenine Phosphoribosyl Transferase Enzyme Deficiency in Renal Transplantation: A Case Report
Corresponding author: Praveen Vijaykumar Malavade, Department of Nephrology, NEPHROURO (NU) Hospitals, Shivamogga, Karnataka, India. E-mail: malvadepraveen3@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Malavade PV. Successful Management of Adenine Phosphoribosyl Transferase Enzyme Deficiency in Renal Transplantation: A Case Report. Indian J Nephrol. 2024;34:383-5. doi: 10.25259/IJN_167_2024
Abstract
Adenine phosphoribosyltransferase (APRT) enzyme deficiency is a rare inborn metabolic error causing an accumulation of 2,8 dihydroxyadenine (DHA), leading to kidney stones and crystal nephropathy. If untreated, it progresses to end stage renal disease (ESRD) with a subsequent risk of crystal nephropathy recurrence post-renal transplantation. Recurrence post-transplant can be prevented, and allograft outcomes can be improved if treatment with an xanthine oxidoreductase (XOR) inhibitor is initiated before or at the time of kidney transplantation. We describe a case involving a 24-year-old male with ESRD, found to have APRT enzyme deficiency during transplant evaluation, successfully managed with pre- and post-transplant XOR inhibitors to prevent recurrence, resulting in a positive allograft outcome.
Keywords
APRT deficiency
XOR inhibitor
Renal failure
Recurrence
DHA crystals

Introduction
Adenine phosphoribosyltransferase (APRT) deficiency (OMIM 102600) is a rare autosomal recessive disorder impacting purine metabolism, linked to the APRT gene on chromosome 16 (16q24).1 It results in the formation of 2,8-dihydroxyadenine (DHA) stones, which are insoluble in urine at normal pH levels, leading to recurrent kidney stones and kidney damage. Clinical manifestations include renal colic, hematuria, urinary tract infections (UTIs), and kidney failure, with symptom onset ranging from infancy to later in life. Approximately 50% of those with APRT deficiency are asymptomatic.
Diagnosis is challenging due to the stones’ radiographic invisibility and the inability of routine tests to distinguish between DHA and uric acid stones. Accurate diagnosis involves stone analysis, identifying DHA crystals in urine or kidney biopsies, and assessing APRT enzyme activity in red blood cells. Treatment includes allopurinol, high fluid intake, and a low-purine diet. Urine alkalinization is ineffective due to DHA’s insolubility across different pH levels [Figure 1]. The disease can recur in transplanted kidneys.
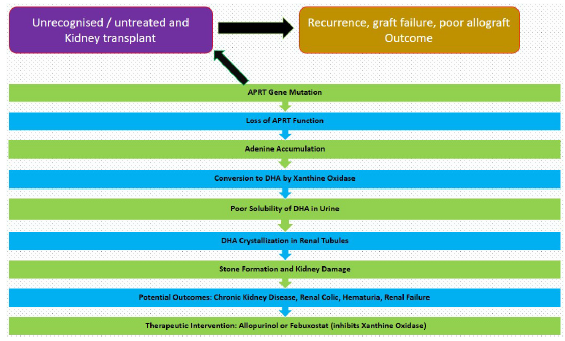
- APRT deficiency. APRT: Adenine phosphoribosyltransferase, DHA: Dihydroxyadenine.
Case Report
A 24-year-old male, born from a consanguineous marriage, first presented with symptoms indicative of renal stones (flank pain and hematuria) at the age of 12. Conservative management was adopted, including increased hydration, dietary modifications, and pain management. No further diagnostic evaluations or follow-up imaging were pursued, and the patient did not report any recurrent stone episodes.
By the age of 20, he was diagnosed with chronic kidney disease. Over the next 4 years, the kidney disease progressed, and he was initiated maintenance hemodialysis (MHD). He came to us for kidney transplantation 2 years later. No significant involvement of other organ systems was observed throughout his clinical course. During the pre-transplant evaluation, the patient was diagnosed with hypothyroidism. The genetic evaluation was initially aimed at identifying possible primary oxalosis. However, the results indicated a homozygous nonsense mutation in exon 3 of the APRT gene (chr16:g.88810545G>A; Depth: 117x), which leads to a stop codon and premature truncation of the protein at codon 67 (p.Arg67Ter; ENST00000378364.8) [Figure 2]. This genetic variant was absent in major genomic databases like the 1000 Genomes, gnomAD (v3.1), and gnomAD (v2.1). However, it showed minor allele frequencies of 0.00076% and 0.00231% in the TopMed and the databases of MedGenome Laboratories, respectively. This mutation had been identified in one other reported case of APRT deficiency.2 Urinalysis was not possible due to anuria.
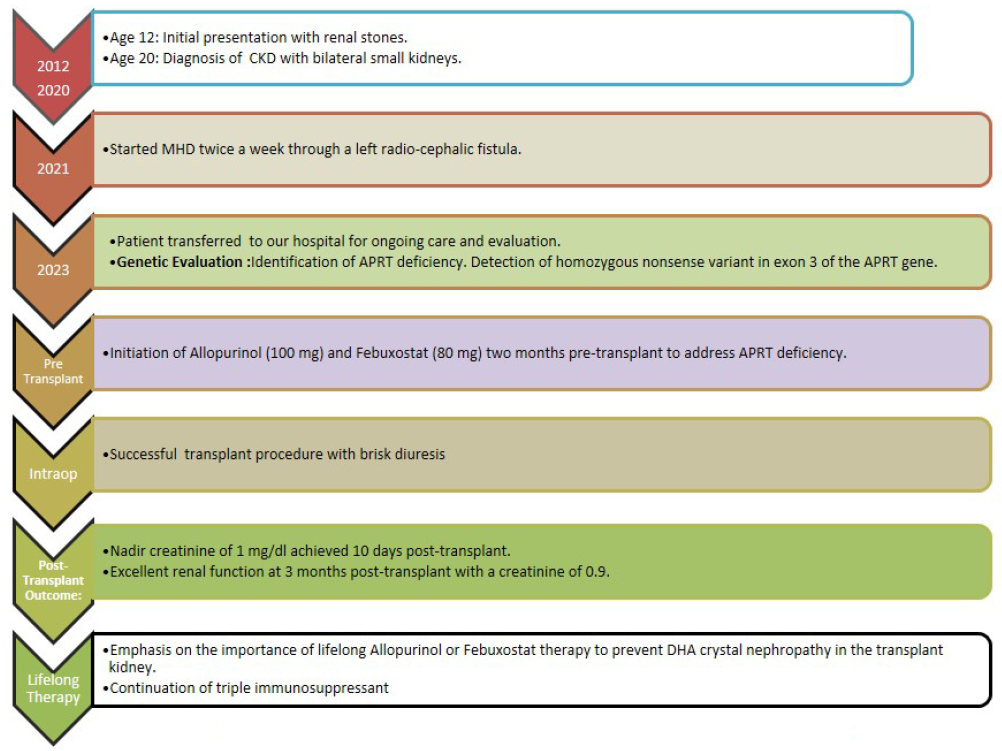
- Journey from renal stones to renal transplantation. CKD: chronic kidney disease, MHD: maintenance hemodialysis, APRT: adenine phosphoribosyl transferase enzyme, DHA: dihydroxyadenine
He was started on allopurinol (100 mg) and febuxostat (80 mg) and received a kidney transplant from his mother. He was induced with basiliximab 20 mg and received standard triple drug immunosuppression. Prompt diuresis was achieved and the serum creatinine levelsdeclined to 1 mg/dl at 10 days. Three months following the transplant, his serum creatinine was 0.9.
Discussion
APRT deficiency, identified in diverse global populations, is often underdiagnosed, leading to missed therapeutic opportunities. The aggressive nature of DHA crystal-induced injury is notably more pronounced in kidney allografts.3 The role of xanthine oxidoreductase (XOR) inhibitors in preventing DHA crystal nephropathy posttransplant is an area of limited data. The largest case series4 documented the disease recurrence in allograft biopsies from patients without adequate pretransplant XOR inhibitor treatment. Use of allopurinol 300 mg or more daily was able to prevent DHA nephropathy in transplant recipients. Patients on XOR inhibitor therapy at the time of kidney transplantation and continued this treatment through the posttransplant period had superior allograft outcomes.
This case represents the second instance in India with a pretransplant APRT deficiency diagnosis and underscores the importance of early diagnosis and initiation of XOR inhibitors like febuxostat before transplantation.
While allopurinol’s safety in CKD stage 3 and beyond is not extensively studied, febuxostat’s safety has been demonstrated in observational studies.5 This case highlights the effectiveness of proactive treatment strategies in preventing DHA nephropathy recurrence posttransplant and the necessity of comprehensive genetic testing for accurate diagnosis in atypical cases, emphasizing the complexities of identifying rare genetic conditions.
Conclusion
This case highlights the effectiveness of proactive treatment strategies in preventing DHA nephropathy recurrence post transplant and the necessity of comprehensive genetic testing for accurate diagnosis in atypical cases, emphasizing th complexities of identifying rare genetic conditions.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
References
- McKusick’s online mendelian inheritance in man (OMIM) Nucleic Acids Res. 2009;37:D793-6.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. J Am Soc Nephrol. 2010;21:679-88.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Kidney disease in adenine phosphoribosyltransferase deficiency. Am J Kidney Dis. 2016;67:431-8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Kidney transplant outcomes in patients with adenine phosphoribosyltransferase deficiency. Transplantation. 2020;104:2120-8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Urate-lowering efficacy and renal safety of febuxostat in patients with hyperuricemia and stage 4–5 chronic kidney disease not yet on dialysis: A meta-analysis of observational studies. Semin Arthritis Rheum. 2022;56:152073.
- [CrossRef] [PubMed] [Google Scholar]




