

Translate this page into:
The Churg-Strauss syndrome: An unusual presentation
Address for correspondence: Dr. V. N. Unni, Department of Nephrology, Amrita Institute of Medical Sciences and Research Centre, Kochi - 682 041, India. E-mail: unnivn@aims.amrita.edu
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The Churg-strauss syndrome (CSS), also referred to as allergic angiitis and granulomatosis is characterized by asthma, peripheral and tissue eosinophilia, extravascular granuloma formation, and vasculitis of multiple organ systems. It is an uncommon disease with an estimated annual incidence of 1-3 per million. Here, we report a case of CSS with glomerulocentric granulomatous reaction with interstitial eosinophils and involvement of retinal vessels.
Keywords
Churg-Strauss syndrome
crescentic glomerulonephritis
glomerulocentric granuloma
retinal vasculitis

Introduction
The Churg-Strauss syndrome (CSS), also called allergic granulomatosis and angiitis, is a multisystem disorder characterized by allergic rhinitis, asthma, and peripheral blood eosinophilia.[1] The most common organ involved is the lung, followed by the skin. The CSS, however, can affect any organ system, including the cardiovascular, gastrointestinal, renal, and central nervous systems.[2] Renal histologic findings observed commonly include vasculitis in arcuate arteries, afferent arterioles, and glomerular capillaries, often with intense eosinophil infiltration. Focal segmental glomerular lesions were present in many cases but rarely does the disease involve majority of glomeruli.[3]
Case Report
A 20-year-old male presented to our institution with history of recurrent rhinosinusitis and bronchial asthma since 2007. He had malaise, anorexia, and weight loss of 10 kg in 6 months. He received antibiotics for his upper respiratory symptoms and underwent functional endoscopic sinus surgery with adenoidectomy and submucosal resection in August 2010. He was found to have mild renal failure in February 2011, when he was evaluated at another center for respiratory tract infection. On examination, he had frontal sinus tenderness; clinical examination was otherwise normal. Investigations showed total leukocyte count 20,500 cu/mm, Hb 12.5 g/dL, platelet count 341,000 cu/mm, peripheral blood smear: normal, serum creatinine 2.48 mg/dL, blood urea 89.7 mg/dL, serum electrolytes and liver function test: normal, urine protein ++, microscopy 5-10 RBCs/HPF. Throat swab culture: normal flora of upper respiratory tract. X-ray chest showed a few bilateral nonhomogenous opacities. ECG, echocardiogram, and ultrasonogram of abdomen were normal. Serum complements (C3 and C4) were normal. P-antineutrophil cytoplasmic antibodies (P-ANCA), C-ANCA, and antinuclear antibody (ANA) were negative. computerized tomographic scan of paranasal sinuses (plain) showed mild mucosal thickening in bilateral maxillary, bilateral ethmoidal, and bilateral frontal sinuses, suggestive of sinusitis. Kidney biopsy showed glomeruli with cellular crescents and many showed granulomatous response around the glomeruli. Many of the crescents [Figure 1a and b] were seen to extend beyond the Bowman's capsule into the interstitium. The capillary tuft showed fibrinoid necrosis. The tubules showed focal nuclear loss, loss of brush border, and simplification. Tubular lumina showed occasional hyaline and WBC casts and tubular atrophy. The interstitium showed patchy edema, moderate to dense mixed inflammatory infiltrate composed of lymphocytes, plasma cells, neutrophils, and numerous eosinophils [Figure 2a]. Ear, nose, and throat specialist opinion was sought; diagnostic nasal endoscopy was performed that showed features suggestive of chronic sinusitis. On ophthalmology evaluation, fluorescein angiography of the right eye showed mild vitreous haze superiorly, leakage around the vessels superiorly, and fuzziness of the vasculature in the superotemporal arcade, signifying vasculitis of retina [Figure 2b].

- (a) Renal biopsy PAS stain (×40) showing glomeruli with granulomatous response around the glomeruli. Many of the crescents are seen to extend beyond the Bowman's capsule (arrow) into the interstitium. (b) PAS stain (×100) showed glomeruli with granulomatous response (arrow) around the glomeruli
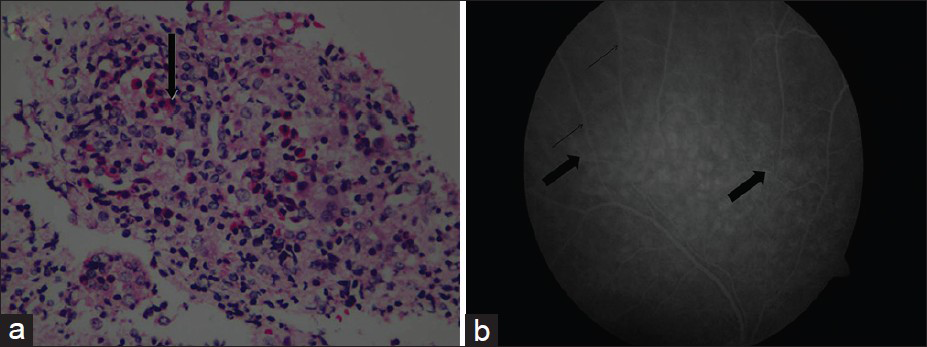
- (a) Renal biopsy showing moderate to dense mixed inflammatory infiltrate composed of lymphocytes, plasma cells, neutrophils, and numerous eosinophils (arrow). H and E stain (×100) (b) Fluorescein angiography of right eye showing mild vitreous haze superiorly, leakage around the vessels superiorly (bold arrow), and fuzziness of the vasculature (thin arrow) in the superotemporal arcade, signifiying vasculitis of retina
In view of history of asthma, features of sinusitis, pulmonary opacities detected radiographically, and kidney biopsy showing fibrinoid necrosis in glomeruli, granuloma, and eosinophils in extravascular areas, a diagnosis of CSS was made. Our patient had four of the six criteria proposed by the American College of Rheumatology for diagnosis of CSS. In our patient, peripheral blood eosinophilia and mononeuritis multiplex were not seen. He was started on prednisolone 1 mg/kg/day and cyclophosphamide 2 mg/kg/day. His serum creatinine level improved during the hospital stay. On follow-up, he was asymptomatic, his renal function improved, and findings on chest X-ray resolved.
Discussion
CSS, a systemic vasculitis, can be distinguished from other necrotizing vasculitis on the basis of clinical and histologic criteria, characterized by the presence of asthma, hypereosinophilia, and necrotizing vasculitis with extravascular granulomas.[4] The characteristic histopathologic feature of CSS is the granulomatous reactions that may be present in the tissues or even within the walls of the vessels themselves. These are usually associated with infiltration of the tissues with eosinophils. This process can occur in any organ in the body. Lung involvement is the predominant one, with skin, cardiovascular system, kidney, peripheral nervous system, and gastrointestinal tract being the other organs involved.[4] Renal involvement is usually not a prominent feature and is usually mild.[1]
The American College of Rheumatology has established six criteria for the diagnosis of CSS.[3] The presence of four or more of these six criteria yields a sensitivity of 85% and a specificity of 99.7% for CSS: asthma (a history of wheezing or the finding of diffuse high pitched wheezes on expiration), eosinophilia of >10% on differential white blood cell count, mononeuropathy (including multiplex) or polyneuropathy, migratory or transient pulmonary opacities detected radiographically, paranasal sinus abnormality, biopsy containing a blood vessel showing the accumulation of eosinophils in extravascular areas.
The disease can occur at any age with the possible exception of infants. The mean age of onset is 48 years, with a female-to-male ratio of 1.2:1.[4] Patients with CSS often exhibit nonspecific manifestations such as fever, malaise, anorexia, and weight loss, which are characteristics of a multisystem disease.[4] Pulmonary involvement in CSS clearly dominates the clinical picture with severe asthmatic attacks and the presence of pulmonary infiltrates, which were present in our patient.
Ocular involvement in CSS is rare. The reported ophthalmologic manifestations include corneal ulcer, uveoscleritis, conjunctival granuloma, orbital inflammatory pseudotumor, amaurosis fugax, retinal artery occlusion, ischemic optic neuropathy, oculomotor nerve palsy, and trochlear nerve palsy.[5] Takanashi et al. classified these ocular manifestations into two types: orbital inflammatory pseudotumor and ischemic vasculitis. They hypothesized that these two groups may represent the two essential characteristics of the disease processes: granulomatosis and angiitis.[6] In our patient, fluorescein angiography of right eye showed mild vitreous haze superiorly, leakage around the vessels superiorly, and fuzziness of the vasculature in the superotemporal arcade, suggestive of vasculitis of retina.
ENT involvement is common in CSS, usually manifesting as allergic rhinitis and chronic rhinosinusitis with or without polyps. Otolaryngologists may play a pivotal role in making an early diagnosis of this disease.[7]
Renal involvement is not regarded as a prominent feature, and its prevalence and severity vary widely.[8] In a study by Sinico et al. of 48 men and 68 women with a mean age of 51.9 years (range 18-86 years), renal abnormalities were present in 31 patients (26.7%). Rapidly progressive renal insufficiency was documented in 16 patients (13.8%), urinary abnormalities in 14 patients (12.1%), and chronic renal impairment in one patient. Of these, 16 patients underwent renal biopsy, out of which 11 showed necrotizing crescentic glomerulonephritis. Other findings were eosinophilic interstitial nephritis, mesangial glomerulonephritis, and focal sclerosis.[9]
The first step in the management of CSS is to assess the severity of the disease. A frequently used system is the “five-factors score” (FFS) based on the presence or absence of five clinical factors: cardiac involvement, gastrointestinal disease, renal insufficiency (plasma creatinine concentration >1.6 mg/dL [141 μmol/L]), proteinuria (>1 g/day), and central nervous system involvement. The presence of each factor is given one point. The FFS ranges from 0 to 2, a score of 0 is given when none of the factors is present, a score of 1 for one factor, and a score of 2 for two or more factors. This scoring system has also been correlated with prognosis.[7]
Peripheral blood eosinophilia (usually 5000-9000 eosinophils/μL) is a strong pointer toward CSS, although levels over 1500 cells/μL (or greater than 10% of the total leukocyte count) should arouse a suspicion of CSS. Eosinophilia, however, is occasionally missed because of rapid spontaneous or glucocorticoid-induced reductions or fluctuations in eosinophil counts. Tissue eosinophilia can still be found in patients in whom peripheral blood eosinophilia is absent.[10] ANCA are found in 40% to 60% of patients with CSS. The majority of ANCA-positive CSS patients (70% to 75%) have antibodies directed against myeloperoxidase with a perinuclear staining pattern (called myeloperoxidase-ANCA or P-ANCA).[11]
Surgical lung biopsy, although not always available, is the gold standard for the diagnosis of CSS. On the contrary, transbronchial lung biopsy is generally not helpful.[5] Radiographic manifestations may remain stable or may rapidly regress with glucocorticoid treatment.
When either skin disease or peripheral neuropathy is present, biopsy of one of those sites is less invasive and often preferred to a lung biopsy. In one study, 15 of 28 patients with a peripheral neuropathy and CSS had evidence of necrotizing vasculitis on a peripheral nerve biopsy.[5]
The primary therapy for CSS involves systemic glucocorticoids. Asthma is additionally treated with inhaled glucocorticoids. Refractory CSS may be responsive to cyclophosphamide, azathioprine, or high-dose intravenous immune globulin, and small numbers of patients have been treated successfully with the combination of systemic glucocorticoids and interferon-alpha.[4]
For all patients with CSS and evidence of systemic vasculitis, systemic glucocorticoid therapy is recommended. Oral prednisone in doses of 0.5-1.5 mg/kg per day is administered for 6-12 weeks or until disease remission is attained, and then gradually tapered. Patients with fulminant disease may require initial therapy with intravenous glucocorticoids. For patients with severe disease manifested by a FFS of 2, addition of cyclophosphamide to systemic glucocorticoid therapy is warranted. Patients with a FFS of 1 (especially with cardiac or central nervous system involvement) should receive cyclophosphamide and systemic glucocorticoids.[6] After induction of remission with cyclophosphamide, a transition to maintenance therapy with azathioprine to sustain the remission can be attempted. Methotrexate is an alternative agent that can be used if azathioprine is not tolerated or is not effective. These drugs are preferred to long-term cyclophosphamide therapy, which is associated with significantly greater toxicity. Immunosuppressive therapy should be continued for 12-18 months. Long-term or indefinite maintenance therapy may be warranted in patients with multiple relapses.[7]
Granulomatosis with polyangiitis (Wegener's), microscopic polyangiitis, and CSS can all affect the lung, although the presence of eosinophilia and asthma is typical of CSS and is not usually seen in the other two.
The American College of Rheumatology proposed four clinical criteria for the classification of granulomatosis with polyangiitis (Wegener's), abbreviated as GPA.
-
Nasal or oral inflammation (painful or painless oral ulcers, or purulent or bloody nasal discharge).
-
Abnormal chest radiograph showing nodules, fixed infiltrates, or cavities.
-
Abnormal urinary sediment (microscopic hematuria or red cell casts).
-
Granulomatous inflammation on biopsy of an artery or perivascular area.
The presence of two or more of these four criteria yielded a sensitivity of 88% and a specificity of 92% for the diagnosis of granulomatosis with polyangiitis.[6]
Monitoring responsiveness to treatment and the development of recurrence is best achieved by following the eosinophil count and erythrocyte sedimentation rate. Persistence of ANCA positivity in CSS may be a marker of an underlying disease process, but does not appear to adequately reflect disease activity and, thus, cannot be used to determine changes in therapy. This was demonstrated in a retrospective study of 53 patients with polyarteritis nodosa or CSS in whom the persistence of ANCA positivity did not correlate with activity of the underlying disease.[12] The type of ANCA seen in CSS is more typically antimyeloperoxidase, whereas in CSS it is more often antiproteinase 3. ANCA are found in 40% to 60% of patients with CSS. The majority of ANCA-positive CSS patients (70% to 75%) have antibodies directed against myeloperoxidase with a perinuclear staining pattern (called MPO-ANCA or P-ANCA).[13]
Late relapses after a successful response to treatment are uncommon. As a result, treatment can be discontinued in most patients. However, premature withdrawal of treatment can result in recurrence. Hypertension should be managed in a standard fashion, but may be difficult to control.[14]
Prognosis
Although the prognosis of patients with CSS is unclear, treatment appears to have significantly decreased mortality. Prior to the use of glucocorticoids, the disease was uniformly fatal, with 50% of untreated patients dying within three months of the onset of vasculitis. In contrast, recent reports suggest a survival rate of greater than 70% at 5 years.[15]
The presence or absence of certain clinical features strongly correlates with subsequent mortality. The following five clinical factors appear to have significant adverse prognostic value in patients with CSS.[11] The presence or absence of the features that make up the FSS has been used to predict survival in CSS (i.e., cardiac involvement, gastrointestinal disease [bleeding, perforation, infarction, or pancreatitis], renal insufficiency (plasma creatinine concentration >1.6 mg/dL [141 micromol/L]), proteinuria (>1 g/day), and central nervous system involvement). Of these, the presence of significant cardiac or gastrointestinal disease appears to be the strongest indicators of poor prognosis.[11]
Conclusion
Although the renal disease in CSS is less common and generally less severe than that of Wegener's granulomatosis and microscopic polyangiitis, some cases might present with crescentic glomerulonephritis and renal dysfunction. We report this case as the patient had characteristic glomerulocentric granuloma on renal biopsy, and involvement of retinal vessels is a rare presentation of this uncommon disease.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol. 1951;27:277-301.
- [Google Scholar]
- Renal involvement in Churg-Strauss syndrome. Nephrol Dial Transplant. 1990;5:161-7.
- [Google Scholar]
- The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis) Arthritis Rheum. 1990;33:1094-100.
- [Google Scholar]
- The vasculitis syndromes. In: Harrison's Principles of Internal Medicine (18th ed). McGraw-Hills: New York; 2011. p. :2785-805.
- [Google Scholar]
- Clinicopathological features of Churg-Strauss syndrome-associated neuropathy. Brain. 1999;122:427-39.
- [Google Scholar]
- The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990;33:1101-7.
- [Google Scholar]
- Orbital inflammatory pseudotumor and ischemic vasculitis in Churg-Strauss syndrome: Report of two cases and review of the literature. Ophthalmology. 2001;108:1129-33.
- [Google Scholar]
- Pauci Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis. In Hepinstall's Pathology of the Kidney. (6th ed). Philadelphia: Lippincott Williams and Wilkins; 2007. p. :643-73.
- [Google Scholar]
- Systemic vasculitis with asthma and eosinophilia: A clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore). 1984;63:65-81.
- [Google Scholar]
- Persistent airflow obstruction in asthma of patients with Churg-Strauss syndrome and long-term follow-up. Allergy. 2009;64:589-95.
- [Google Scholar]
- Persistence of antineutrophil cytoplasmic antibodies (ANCA) in asymptomatic patients with systemic polyarteritis nodosa or Churg-Strauss syndrome: Follow-up of 53 patients. Clin Exp Rheumatol. 1995;13:193-8.
- [Google Scholar]
- Churg-Strauss syndrome: Serum markers of lymphocyte activation and endothelial damage. Arthritis Rheum. 1998;41:445-52.
- [Google Scholar]
- Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med. 2005;143:632-8.
- [Google Scholar]
- Churg-Strauss syndrome.Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore). 1999;78:26-37.
- [Google Scholar]




