

Translate this page into:
An unusual case of nephrotic syndrome
Address for correspondence: Dr. M. Sahay, Department of Nephrology, Osmania General Hospital and Medical College, 6-3-852/A, Ameerpet, Telangana - 500 016, Hyderabad, India. E-mail: drmanishasahay@gmail.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Nephrotic syndrome can be rarely due to inherited disorders of enzymes. One such variety is lecithin cholesterol acyltransferase deficiency. It leads to accumulation of unesterified cholesterol in the eye and other organs. We report a case of nephrotic syndrome with cloudy cornea and hypocholesterolemia with foam cells and lipid deposits on renal biopsy. Awareness about this rare disease may help in the early institution of specific measures to prevent progression to end-stage renal disease.
Keywords
Corneal deposits
lecithin cholesterol acyl transferase
nephrotic syndrome
renal foam cells

Introduction
Nephrotic syndrome (NS) is a common renal disorder in adults. Secondary causes include drugs, collagen vascular diseases, diabetes, malignancy, etc. Genetic causes are also known and may be due to inherited defects in podocyte proteins or due to enzyme deficiency. One such variety is NS due to deficiency of lecithin cholesterol acyl transferase (LCAT) enzyme. It is seen rarely and only 60 cases have been reported so far. It is often associated with cloudy cornea. We report a case with nephrotic syndrome and cloudy cornea and foam cells and lipid deposits on renal biopsy.
Case Report
A 28-year-old male presented with a history of frothy urine and swelling of feet of 2 months duration. He complained of blurring of vision, which was rectified by glasses. He did not have any features of systemic disease. There was no hypertension or diabetes mellitus. He was born of nonconsanguinous marriage and was the youngest of three children. No other members in the family were affected. Examination revealed pedal edema, cloudy cornea and arcus juvenilis. Examinations of respiratory system, cardiovascular system and nervous system were normal. Investigations showed urine specific gravity of 1.015, protein 3+, and blood 3+. Microscopy showed 1–2 pus cells/high power field and few epithelial cells. There were 30–35 red blood cells per high power field. Twenty-four hour urine protein was 5732 mg. Fasting blood sugar was 104 mg/dl, postprandial sugar 117 mg/dl, serum creatinine 1.1 mg/dl, sodium143 mmol/l, potassium 3.8 mmol/l, low-density lipoprotein cholesterol 59 mg/dl, triglycerides 253 mg/dl, total cholesterol 122 mg/dl, very-low-density lipoprotein 51 mg/dl and high-density lipoprotein (HDL) 12 mg/dl. Ultrasound examination showed right kidney measuring 12.1 × 4.7 and left kidney measuring 12.2 cm × 5.5 cm. Eye examination shows cloudy cornea and arcus juvenilis [Figure 1]. Renal biopsy [Figure 2] showed enlarged glomeruli, with nodular deposits in the mesangium with irregularly thickened basement membrane, foam cell infiltrate of macrophages containing lipid material in capillaries and mesangial areas. Tubulo interstitial compartment and blood vessels were normal. Stain for amyloid was negative. Immunofluorescence revealed insignificant mesangial deposits of IgM and C3c. Electron microscopy showed diffuse effacement of podocyte foot processes, capillary loop and mesangial regions were distended by electron lucent lipid material containing electron dense proteinaceous matrix suggestive of LCAT deficiency. The patient was started on ramipril 10 mg/day. There was partial remission of proteinuria with stable renal functions at 1-year follow-up.
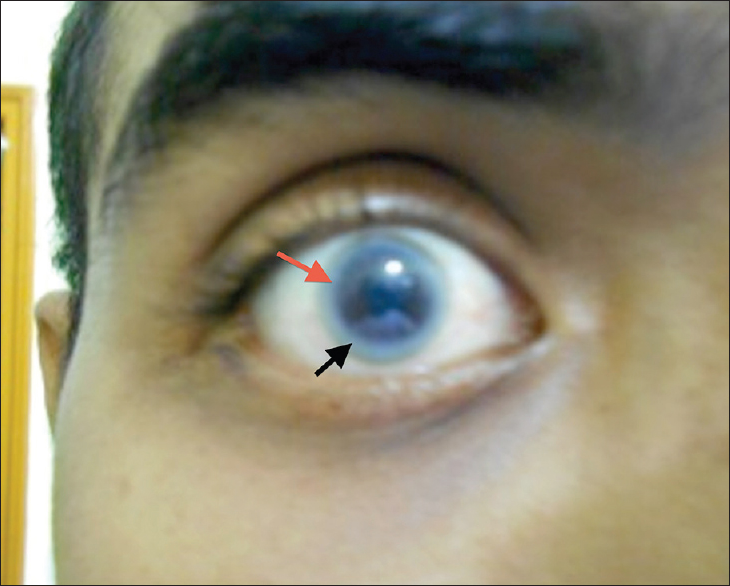
- Cloudy cornea (red arrow), arcus junvenilis (black arrow)

- (a) Renal histology with foam cells in mesangium on light microscopy (arrow) (H and E, ×40), (b) renal histology with electron dense mesangial deposits (arrow)
Discussion
The patient presented with nephrotic syndrome with cloudy cornea. The serum cholesterol levels were low which is unusual in a patient with nephrotic syndrome. Renal histology showed foam cells and lipid deposition in mesangium and glomerular capillaries. Cloudy cornea may be a feature of other nephrotic states and isolated foam cells may be seen in other renal diseases with massive proteinuria. However, the combination of nephrotic syndrome, cloudy cornea, foam cells with lipid deposits in glomeruli and very low serum HDL are typical of complete LCAT deficiency. LCAT is synthesized in the liver and catalyzes the formation of cholesterol esters.[1] Deficiency of this enzyme leads to accumulation of unesterified cholesterol in the cornea leading to cloudy cornea and organomegaly.[23] Partial deficiency leads to fish eye disease, which is characterized by cloudy cornea without any systemic involvement. LCAT deficiency is a rare autosomal recessive disorder caused by mutations in LCAT gene on long arm of chromosome 16.[124] Hemolytic anemia, and lymphadenopathy have also been described but were not seen in our patient. Atherosclerosis is rare but is reported.[5] Plasma LCAT activity is low and may be helpful for diagnosis in atypical cases. Treatment consists of ACEI, and dietary fat restriction. The disease is slowly progressive and the patient may eventually need renal replacement therapy. The patient has diminished vision and is using glasses. He may benefit from corneal transplantation if his vision is drastically reduced. LCAT enzyme supplementation is becoming available and may prevent end stage renal disease in such patients.[6]
Source of Support: Nil
Conflict of Interest: None declared.
References
- The molecular pathology of lecithin: Cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res 1997:191-205.
- [Google Scholar]
- Lecithin-cholesterol acyltransferase (LCAT) deficiency without mutations in the coding sequence: A case report and literature review. Clin Nephrol. 2011;76:323-8.
- [Google Scholar]
- High prevalence of mutations in LCAT in patients with low HDL cholesterol levels in the Netherlands: Identification and characterization of eight novel mutations. Hum Mutat. 2011;32:1290-8.
- [Google Scholar]
- Familial plasma lecithin: Cholesterol acyltransferase deficiency. Scand J Clin Invest. 1967;20:231-43.
- [Google Scholar]
- Genetic lecithin: Cholesterol acyltransferase deficiency and cardiovascular disease. Atherosclerosis. 2012;222:299-306.
- [Google Scholar]
- Familial LCAT deficiency: From renal replacement to enzyme replacement. Neth J Med. 2013;71:29-31.
- [Google Scholar]




