

Translate this page into:
An unusual cause of gross hematuria and renal dysfunction in a young male
Address for correspondence: Dr. Manish Rathi, Department of Nephrology, Post Graduate Institute of Medical Education and Research, Chandigarh 160 012, India. E-mail: drmanishrathi2000@yahoo.co.in
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Monoclonal immunoglobulin deposition disease (MIDD) is an uncommon disease with a peak incidence between the 5th and 6th decades of life. It is characterized by non-fibrillar, Congo red negative deposition of monoclonal immunoglobulins in various organs, including in the kidneys. MIDD can be of three types depending on the composition of the deposits, and includes light chain deposition disease (LCDD), heavy chain deposition disease and light and heavy chain deposition disease, of which LCDD is the most common. Renal involvement is a universal finding in MIDD, and is in the form of renal insufficiency, microscopic hematuria and nephrotic range proteinuria. Gross hematuria is a rare occurrence. Renal biopsy usually shows nodular sclerosing glomerulopathy on light microscopy and diffuse linear staining of glomerular and tubular basement membrane on immunofluorescence microscopy. We report a young male who presented with rapidly progressive renal failure and gross hematuria and was diagnosed as LCDD with nodular glomerulopathy and crescents on renal biopsy.
Keywords
Monoclonal immunoglobulin deposition disease
multiple myeloma
rapidly progressive renal failure

Introduction
Monoclonal immunoglobulin deposition disease, (MIDD) is a systemic disease characterized by non-fibrillar, Congo red negative deposition of monoclonal immunoglobulins in the various organs of the body. MIDD is of three types depending on the composition of the deposits: Light chain deposition disease (LCDD), heavy chain deposition disease (HCDD) and light and heavy chain deposition disease (LHCDD). Of these three types, LCDD is the most common, comprising about 80% of the cases.[1] LCDD is usually seen in association with multiple myeloma or other lymphoproliferative disorders, and presents with renal involvement in the form of proteinuria and renal insufficiency.[2] Microscopic hematuria is seen in 60% of the cases, while gross hematuria is rare.[3] We hereby present a young male who developed gross hematuria and rapidly progressive renal failure and was diagnosed to have non-myeloma related LCDD.
Case Report
A 31-year-old male presented with 1 week history of anorexia, headache, and gross hematuria. He did not have history of fever, urinary tract infection, stone disease, rash, arthralgia, night sweats, and weight loss, nor was there any significant past history. He was a non-smoker and non-alcoholic, and family history was not contributory. On examination, his blood pressure was 180/100 mmHg, heart rate was 100/min and he had mild pallor. Systemic examination was normal and fundus examination did not reveal any changes of hypertensive retinopathy.
Investigations revealed normal hemoglobin (11.5 g/l), total leukocyte count (9500/dl) and platelet count (2.4 × 106/dl), but increased serum creatinine (4.8 mg/dl). Urine examination revealed proteinuria (4+) and dysmorphic RBCs (>80% of RBCs), and his 24-h urine protein excretion was 3.8 g. For evaluation of gross hematuria, he underwent cystoscopic examination, ultrasonography and non-contrast computerized tomography of the genitourinary tract, which were normal. Anti-nuclear antibodies, anti-neutrophil cytoplasmic antibodies, anti-glomerular basement membrane antibodies and serum cryoglobulins were negative, so were hepatitis B surface antigen, anti-hepatitis C antibody and human immunodeficiency virus. His renal function further deteriorated, requiring initiation of hemodialysis, and subsequently, a kidney biopsy was performed. The kidney biopsy specimen contained nine glomeruli, and consisted of both cortex and medulla. Light microscopy examination of the biopsy revealed that all the glomeruli showed mesangial expansion, hypercellularity and mesangial nodules. Cellular crescents were present in two glomeruli along with focal glomerular basement membrane thickening. The tubulointerstitial compartment showed fibrosis and atrophy involving <30% of the biopsy area, while the blood vessels were normal [Figure 1]. The mesangial nodules were periodic acid-schiff (PAS) positive but Congo red negative. Immunofluorescence examination revealed 2+ to 3+ diffuse linear staining for kappa light chains in mesangium and tubular basement membrane [Figure 2]. Electron microscopy examination showed powdery electron-dense material in the lamina rara interna of the glomerular basement membrane as well as on the outer aspect of the tubular basement membrane [ a and b]. With these findings, a diagnosis of LCDD was made and he was investigated further.

- Light microscopy showing mesangiocapillary pattern of injury (a), with PAS positive nodules and crescent formation (b)
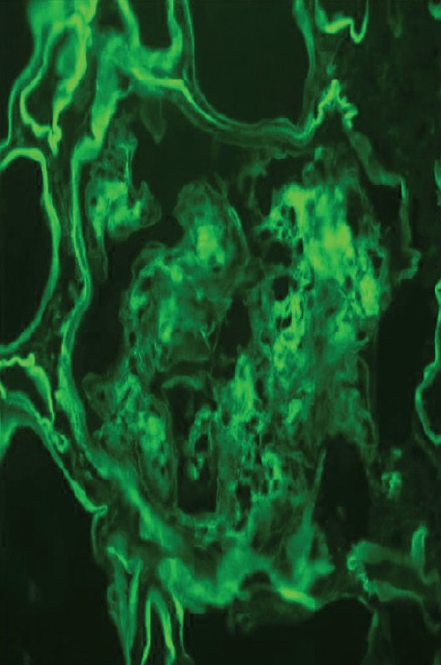
- Immunofluorescence showing positivity with kappa-light chain in mesangium and tubular basement membrane
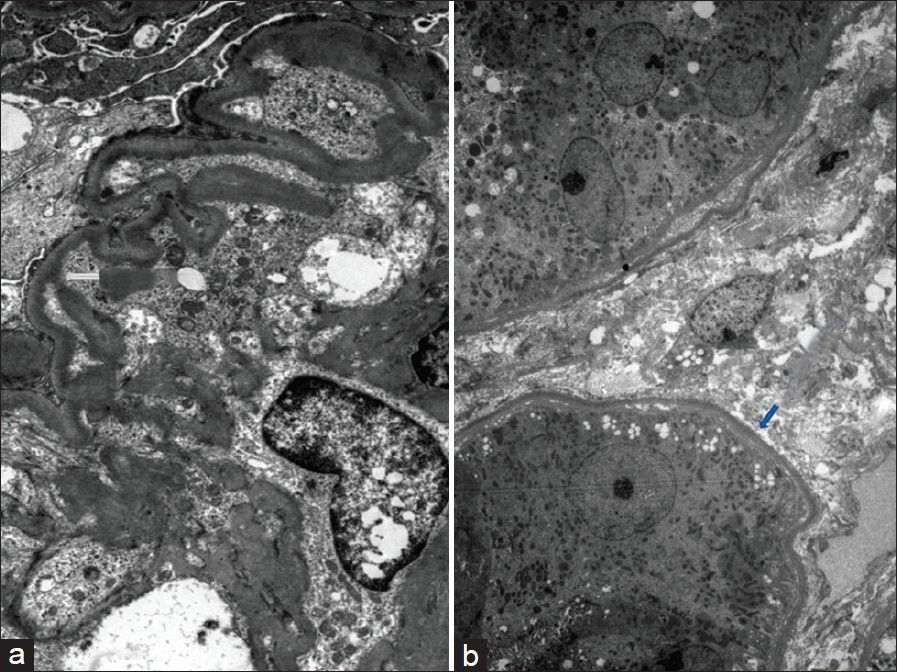
- Electron microscopy showing powdery electron-dense material in lamina rara interna of glomerular basement membrane (a) and on the outer aspect of tubular basement membrane (b) (uranyl acetate, × 1080)
Serum electrophoresis was performed, which revealed a monoclonal band in the gamma region that, on immunofixation, was confined to the kappa and IgG regions. Serum kappa free light chain level was elevated and bone marrow examination revealed 8% plasma cells. Thus, the LCDD was thought to be non-myeloma related. He was started on bortezomib, thalidomide and dexamethasone regimen. After four cycles, his hematuria settled; however, renal functions did not improve. Repeat serum immunofixation was negative. He was initiated on maintenance hemodialysis on which he is doing well since the last 3.5 years and is planned for deceased donor renal transplantation.
Discussion
MIDD is a rare disease characterized by deposition of monoclonal light and/or heavy chains of immunoglobulins in various tissues. It has three subtypes: LCDD, HCDD, and LHCDD, of which LCDD is the most common, accounting for about 80% of the cases.[12] All the three forms have similar clinical and histological features and can be differentiated only by estimation of light and heavy chain levels in serum or by immunological examination of the tissues. MIDD can be found in up to 5% of myeloma cases on autopsy, while in another study, it was seen in 0.33% of all renal biopsies done at a single center over an 18-year-period.[1] Myeloma is the underlying cause of MIDD in 31-65% of the cases. By immunofixation, a monoclonal protein is seen in 69% of the patients in serum and 78% in urine, while up to 17% of the patients do not have monoclonal proteins in either serum or urine.[23] The diagnosis of LCDD often precedes other clinical features of dysproteinemia and commonly is the presenting feature that leads to diagnosis of multiple myeloma.[4]
MIDD is usually seen in the 5th-6th decade of life; however, up to one-third of the cases can be <50 years of age.[3] The manifestations depend on the organs affected by immunoglobulin deposition. Patients usually have cardiac, neural, hepatic and renal involvement, of which renal involvement is the most common.[12] Non-selective proteinuria occurs in >90% of the cases;[3] however, proteinuria may be absent during the early phase of disease and it does not correlate with the presence of nodular glomerulosclerosis.[5] Nephrotic range proteinuria can be seen in up to 23-57% of the patients,[15] while microcytic hematuria is seen in around 60% of the patients and gross hematuria is seen rarely.[3] At presentation, 80% of the patients are hypertensive and >90% have renal insufficiency (serum creatinine >1.2 mg%). However, dialysis requirement at presentation is seen in only 16%.[3] Patients with a combination of both LCDD and cast nephropathy usually have more severe disease and renal insufficiency as compared with cases with LCDD alone.[23] Other symptoms include hepatomegaly, mild alteration in liver functions, cardiomegaly, conduction abnormalities, heart failure and peripheral neuropathy.[1]
The pathological features are dominated by deposition of monoclonal immunoglobulin in various organs. In kidneys, these deposits are mainly in the glomerular basement membrane and tubular basement membrane. The light microscopy examination of renal biopsy commonly shows acellular, nodular glomerulosclerosis.[16] The glomeruli are enlarged with diffuse, nodular expansion of the mesangial matrix along with mild cellularity. These nodules are eosinophilic and PAS positive, but negative with Jones’ silver methenamine and Congo red stains. Nodular glomerulosclerosis of LCDD in light microscopy appears similar to nodules found in other glomerular diseases, including diabetic nephropathy, membranoproliferative glomerulonephritis, amyloidosis and fibrillary glomerulonephritis. However, nodules of MIDD do not stain with Congo red and silver stain, which differentiates it from amyloidosis (Congo red positive) and diabetic nephropathy (Jones methenamine positive), respectively.[1] The glomerular basement membrane may appear thickened, bright, and rigid. Other rare manifestations include immune complex-mediated disease and minimal change glomerulopathy if biopsy is done at an early stage of the disease.[78] The extraglomerular changes include “ribbon like” thickening of tubular basement membrane and the vessel wall.[8] The interstitium shows variable degree of inflammatory infiltrate and interstitial fibrosis. Patients with gross hematuria may have presence of RBC casts in the tubules due to interaction of filtered RBC with Tamm-Horsfall protein. These RBC casts can themselves generate an inflammatory reaction, leading to damage to the tubulointerstitium.[9] Up to one-third of the patients with MIDD secondary to multiple myeloma may have coexisting light microscopic features of both immunoglobulin deposition and cast nephropathy.[1] Presence of crescents in MIDD has been described mainly in association with alpha heavy chain disease[10] and less commonly, with LCDD.[11]
Immunofluorescence microscopy or immunohistochemistry is the gold standard for diagnosis of MIDD. Presence of monoclonal immunoglobulin deposits in glomerular and tubular basement membrane is pathognomonic of MIDD.[1] In cases of LCDD, kappa light chain deposition predominates over lambda light chain (κ:λ = 4:1). Ultra structural examination shows flocculent to granular to powdery electron-dense material in glomerular basement membrane (100%), mesangium (96%), tubular basement membrane (96%), interstitium (18%), and vascular basement membrane (78%).[8]
The goals of treatment in MIDD are to control proliferation of plasma cells, preserve renal function and improve survival by using chemotherapeutic agents and autologous hematopoietic cell transplantation. Treatment of non-myeloma LCDD is not clear; chemotherapy with alkylating agents and steroids have shown modest results.[1] Bortezomib has been tried with success in patients with LCDD along with other chemotherapeutic agents.[12]
With therapy, 42% of the LCDD patients had either stabilization or improvement in renal function, 8% had worsening renal function (>50% increase in serum creatinine from baseline) and 50% progressed to end-stage kidney disease.[23] Age and serum creatinine at presentation are important predictors of renal survival, while age and coexisting myeloma are predictors of overall survival.[13] Renal transplantation is associated with recurrence of the disease post-transplant. Renal recurrence is seen in about 70% of the cases, but is not always associated with rapid graft loss. The median time to reach end-stage renal disease was 33.3 months.[14] Therefore, renal transplantation could be done to improve quality of life, but should be only carried out after achieving complete remission with chemotherapeutic agents and/or hematopoietic cell transplantation.
The highlight of our case is that he was a young person presenting with gross hematuria and rapidly progressive renal failure with renal biopsy showing crescent formation along with mesangial nodules. A cursory examination of such biopsies can be labeled as mesangio-capillary glomerulonephritis with crescent formation. Thus, a detailed evaluation with immunofluorescence and electron microscopy, along with a high index of suspicion, is required to properly diagnose such cases.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Immunoglobulin light (heavy)-chain deposition disease: From molecular medicine to pathophysiology-driven therapy. Clin J Am Soc Nephrol. 2006;1:1342-50.
- [Google Scholar]
- Renal monoclonal immunoglobulin deposition disease: The disease spectrum. J Am Soc Nephrol. 2001;12:1482-92.
- [Google Scholar]
- Renal monoclonal immunoglobulin deposition disease: A report of 64 patients from a single institution. Clin J Am Soc Nephrol. 2012;7:231-9.
- [Google Scholar]
- Monoclonal immunoglobulin light chain-related renal diseases. Semin Nephrol. 1993;13:324-41.
- [Google Scholar]
- Light chain deposition disease: A model of glomerulosclerosis defined at the molecular level. J Am Soc Nephrol. 2001;12:1558-65.
- [Google Scholar]
- Nonamyloidotic monoclonal immunoglobulin deposition disease. Light-chain, heavy-chain, and light-and heavy-chain deposition diseases. Hematol Oncol Clin North Am. 1999;13:1235-48.
- [Google Scholar]
- Expanding the pathologic spectrum of light chain deposition disease: A rare variant with clinical follow-up of 7 years. Mod Pathol. 2005;18:998-1004.
- [Google Scholar]
- Morphologic heterogeneity of renal light-chain deposition disease. Ultrastruct Pathol. 2008;32:17-24.
- [Google Scholar]
- AKI associated with macroscopic glomerular hematuria: Clinical and pathophysiologic consequences. Clin J Am Soc Nephrol. 2012;7:175-84.
- [Google Scholar]
- Renal crescentic alpha heavy chain deposition disease: A report of 3 cases and review of the literature. Am J Kidney Dis. 2011;58:621-5.
- [Google Scholar]
- Treatment of light chain deposition disease with bortezomib and dexamethasone. Haematologica. 2009;94:300-2.
- [Google Scholar]
- Long-term follow-up and response to chemotherapy in patients with light-chain deposition disease. Am J Kidney Dis. 1992;20:34-41.
- [Google Scholar]
- Long-term outcome of renal transplantation in light-chain deposition disease. Am J Kidney Dis. 2004;43:147-53.
- [Google Scholar]




