

Translate this page into:
ANCA-Associated Vasculitis: Practical Issues in Management
Corresponding Author: Prof. Alan David Salama, UCL Department of Renal Medicine, Royal Free Hospital, London NW3 2PF, UK. E-mail: a.salama@ucl.ac.uk
-
Received: ,
Accepted: ,
How to cite this article: Roper T, Salama AD. ANCA-Associated Vasculitis: Practical Issues in Management. Indian J Nephrol 2024;34:6-23. doi: 10.4103/ijn.ijn_346_23
Abstract
ANCA associated vasculitides are multi-system autoimmune diseases which are increasing in prevalence. In this review we will discuss the clinical manifestations and review the management options. We highlight the various trials of induction and maintenance therapy and discuss the areas of unmet need. These include understanding which patients are at highest risk of relapse, clinical adaptation of improved biomarkers of disease activity and tools to discuss long term prognosis
Keywords
Anti neutrophil cytoplasm antibody
glucocorticoids
granulomatosis with polyangiitis
microscopic polyangiitis
pathogenesis
treatment
vasculitis

Introduction
Vasculitis or inflammation of blood vessel walls can affect vessels of any size throughout the body. The Chapel Hill Consensus Conference (CHCC) criteria 2012 subcategorize these conditions according to the size of vessels they most commonly affect.1 Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV) are a group of rare systemic autoimmune conditions most commonly observed to affect small blood vessels. Three main conditions fall under the AAV umbrella and are defined according to clinical phenotype; granulomatosis with polyangiitis (GPA, formerly Wegener’s granulomatosis); microscopic polyangiitis (MPA), and eosinophilic GPA (EGPA, formerly Churg– Strauss syndrome). A fourth condition, renal-limited vasculitis (RLV), is also classified as an AAV; however, it lacks several of the systemic features associated with the other conditions and can be thought of as a renal-limited variant of MPA.
Different types of AAV show overlap in many of their clinical features (see below); however, the hallmark unifying features for this group of conditions include necrotizing inflammation of small blood vessel walls, absence of immune deposits (termed pauci-immune) on histological analysis, and the presence of circulating antibodies to neutrophil protein antigens. Two such target ANCA antigens have been identified to date, proteinase 3 (PR3) and myeloperoxidase (MPO). PR3-ANCA is more commonly associated with GPA, whereas MPO-ANCA is more often found in those with MPA and RLV. Around 10–20% of cases are ANCA-negative.2,3 Other antibodies targeting neutrophil and endothelial antigens have been found in some cohorts; however, their role in pathogenesis remains uncertain (e.g., anti-moesin, anti-neutrophil elastase, anti-LAMP2).
In the era before effective immunosuppression (IS), 1-year mortality from GPA was >80%.4 Thanks to huge advances in therapeutics that have been made over the last 30 years, survival is now >80% at 1 year and 60–80% at 5 years.5 In this review, we aim to provide an up-to-date overview of the current recommendations in the management of AAV, with a focus on GPA, MPA, and RLV.
Epidemiology
AAV are rare conditions, the incidence of which has been shown to be increasing since first published in the 1980s.6 Most recently, studies describe a combined incidence of 13–20 per million population per year across Europe, North America, and Australasia. The increase in the incidence observed is likely related, in part, to the introduction of routine ANCA serological testing, clearer classification criteria, and an increase in clinician’s awareness of these conditions. In parallel, the combined prevalence of AAV has also been on the rise and is now estimated to be between 30 and 218 per million population globally.7 The rise in prevalence is thought to result from not only the increase in incidence and decrease in diagnostic delay but also from improved survival that has been accompanied by better recognition, and the advent of improved treatment options for these conditions.
The prevalence of GPA and MPA, and associated ANCA subtype serology, differs according to the geographical location. GPA and PR3-ANCA are the more common subtypes identified in populations from Europe and North America, as well as in those from latitudes further from the equator.8 MPA and MPO-ANCA, in contrast, are more often observed in Asian populations, including those living in China and Japan,9,10 as well as those living closer to the equator (Southern Europe and Northern New Zealand).11 Data reporting differences in the incidence between Caucasian and minority ethnic groups within a specific population are variable, with recent reports suggesting no difference in the incidence between minority ethnic groups within a mixed population.12
The majority of studies demonstrate an overall slight male predominance without significant differences in outcomes between the sexes.13,14 There is an increase in the incidence with increasing age, with a peak age of disease onset in those aged 65–74 years.7,15
Pathogenesis
ANCA has been shown to be pathogenic in both in vitro and pre-clinical studies.16-18 The exact reason for the development of these autoimmune antibodies, however, remains unclear and is likely to involve a complex interplay of environmental and genetic risk factors, especially susceptible HLA alleles, alongside a maladaptive immune response.
Environmental factors –the peak age of AAV disease onset in older patients suggests a possible accumulation of environmental risk factors throughout an individual’s lifetime. Several environmental factors have been investigated and demonstrated to be associated with an increased risk of AAV, including exposure to different microparticles, infections, and drugs.
The aforementioned increased prevalence of different types of AAV according to geographical location, most notably latitude, strongly suggests a potential role for environmental factors in the genesis of AAV. Seasonal variations in disease onset have also been reported. Although data are conflicting, some studies suggest an increase in the incidence of GPA in autumn and winter months and MPA in spring and summer months, although issues around the time of disease onset versus clinical presentation make these analyses quite complex.19,20 The exact reason for these geographical and seasonal associations remains unclear. Several studies have attempted to determine potential causes and have concluded that contributing factors may include differences in climate, UV radiation exposure, other environmental exposures, as well as ethnic backgrounds.21 Without understanding exactly what impact these factors have on an individual’s immune response, however, it is difficult to prove causality.
Inhalation of silica dust has been linked to AAV in several cohort studies. A systematic review and meta-analysis demonstrated a positive association between exposure to silica dust and the development of AAV, amongst other autoimmune conditions.22 This association has been demonstrated in those with increased occupational exposure to silica dust (e.g., farming, stone work, mining, construction, and glass work), although it is important to consider the potential role of other microparticles that can be inhaled in these settings (e.g., asbestos, pesticides, organic solvents, hydrocarbons, and heavy metals). Further in support of the association of silica dust inhalation with AAV is the increased incidence of AAV in different regions of Japan following two major earthquakes, both of which were found to be associated with an increase in silica dust following the collapse of buildings and from debris left by tsunamis.23
Bacterial infections have been shown to be associated with both disease onset and relapse in AAV. Staphylococcus aureus has been most frequently associated with AAV, with nasal carriage more frequent in GPA patients and linked to increased relapse rates.24 The reason for this association remains poorly understood; however, it is thought to involve either molecular mimicry or increased exposure to PR3 following the induction of an inflammatory response by bacteria. Recent work using network-based analysis has suggested a direct interaction between S. aureus and dysregulated pathways found in GPA, with potential therapeutic implications.25 Additionally, a few viruses have been implicated as triggers for AAV, although the links between these infections and the occurrence of disease remain weak.26
Several drugs, including vaccines, have been associated with the development of AAV. Therapeutic agents such as propylthiouracil, hydralazine, and minocycline have been linked to the development of symptoms of AAV and positive serum anti-PR3 and -MPO antibodies.27,28 Reports of AAV developing after the use of immune check-point inhibitors (anti-PD-1, anti-PDL-1, and anti-CTLA4) to treat malignancy have recently emerged and are presumed to be due to the stimulation of autoreactive T and B cells.29,30 Both influenza and, more recently, COVID-19 vaccines have been linked to the development of de novo and recurrent AAV.31,32 Illicit drugs are also associated with the development of AAV, the most recognized of these being cocaine (often contaminated with levamisole).33 The exact mechanism by which agents are involved in the development of ANCA antibodies remains unknown but may involve the development of autoimmune-inducing metabolites and polyclonal B cell stimulation.
Genetic factors
Genetic associations with AAV vary with different ANCA serological subtypes (PR3 and MPO) and clinical phenotypes (GPA and MPA). Genome-wide association studies (GWAS) have demonstrated single nucleotide polymorphisms (SNPs) in both human leucocyte antigen (HLA) and non-HLA regions. SNPs in genes encoding HLA-DP, alpha 1-antitrypsin (SERPINA1), a serine protease inhibitor for which PR3 is a substrate, and proteinase 3 (PRTN3) have been linked to those with PR3-AAV and GPA. SNPs in HLA-DQ have been linked to those with MPO-AAV and MPA.34,35 Importantly, a recent meta-analysis showed no genetic overlap between PR3-ANCA and MPO-ANCA AAV.36
Clinical Features
GPA, MPA, and RLV are defined by clinical phenotype and histological findings. Although distinct conditions, GPA and MPA share many clinical features of systemic vasculitis, including; fever, malaise, weight loss, myalgia, and arthralgia, which develop over weeks to months. Organ-specific features tend to develop later on and this can be why there is often a diagnostic delay associated with these conditions and variation in clinical classifications. The relative frequency of organ-specific features of different AAV subtypes can highlight differences in presentation and is summarized from a number of different series, although the referral pathway will skew the proportions of patients with different features [Table 1].1,37,38
GPA
While all types of AAV result from necrotizing inflammation of small blood vessels, GPA (and EGPA) is unique in that this inflammation is associated with granuloma formation. In addition to the non-specific features of systemic vasculitis, GPA most commonly presents with respiratory, renal, ear, nose and throat (ENT), and ocular manifestations. Cough, shortness of breath, and hemoptysis can be a sign of respiratory tract involvement, which can include the presence of lung nodules, cavitating lung lesions, pulmonary capillaritis, and varying degrees of alveolar hemorrhage [Figure 1]. Renal involvement is frequently asymptomatic; however, on testing, those affected may have hematuria, proteinuria, or a rise in serum creatinine (sCr) from baseline values. Kidney biopsy may demonstrate a pauci-immune crescentic glomerulonephritis (GN) of varying severity [Figure 2a]. ENT presentations are broad and include nasal crusting, mucosal ulceration leading to nose bleeds, nasal polyps, destruction of nasal cartilage, eventual saddle nose deformity, hearing loss, otitis media, chronic rhinitis, sinusitis, and laryngitis. Subglottic stenosis remains an important and life-threatening manifestation of GPA and may be associated with both PR3-ANCA and less frequently MPO-ANCA. Eye involvement can present with the inflammation of the anterior or posterior chambers of the eyes, from conjunctivitis, keratitis and uveitis to episcleritis. Orbital and retro-orbital granulomatous masses (pseudo-tumors) can form causing eye pain, proptosis, and diplopia [Figure 3]. Neurological involvement can result in mononeuritis multiplex, which can lead to optic neuritis, sensorineural hearing loss, and peripheral neuropathy. Cutaneous manifestations arise from the inflammation of dermal capillaries that can produce a purpuric or petechial rash, and less commonly ulceration and painful skin lesions.
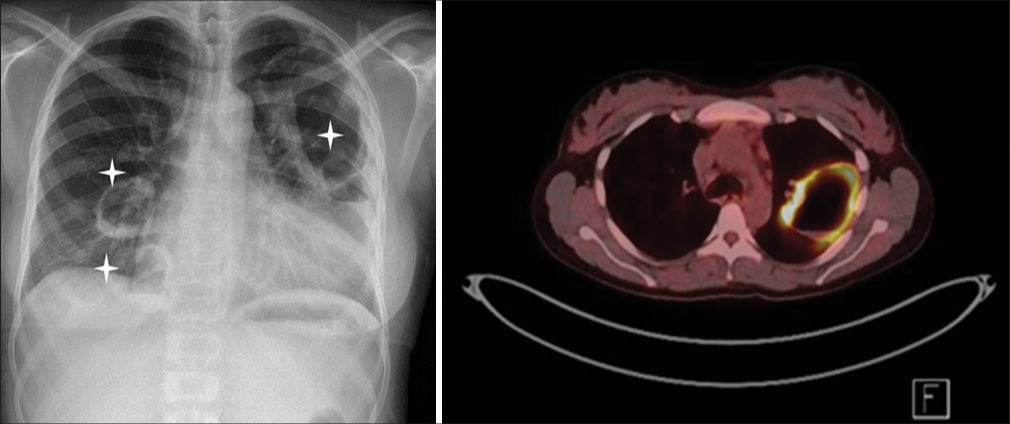
- Cavitating granulomatous lung lesions in a GPA patient observed on plain X-ray (highlighted by an Asterix) and on an FDG-PET.
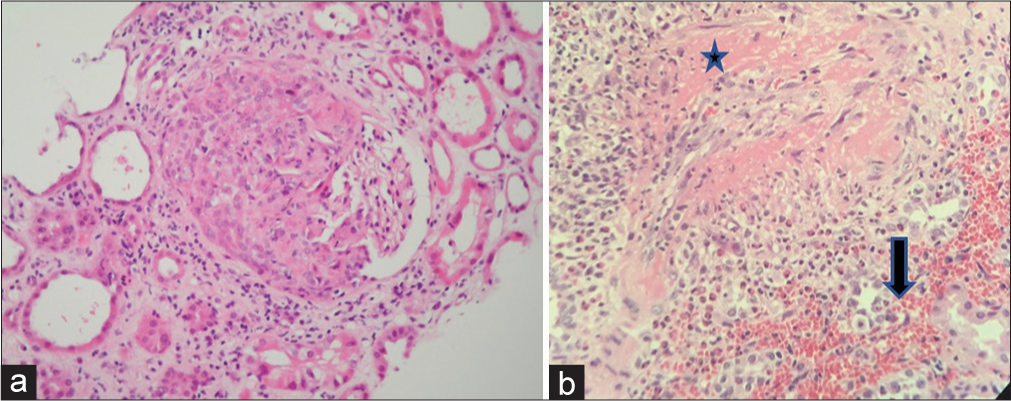
- (a) High power of a single glomerulus showing a crescentic change from a kidney biopsy of a patient with microscopic polyangiitis (H and E ×400), (b) extraglomerular vessel fibrinoid necrosis (Asterix) and interstitial hemorrhage (large arrow) in a kidney biopsy from a patient with granulomatosis with polyangiitis.
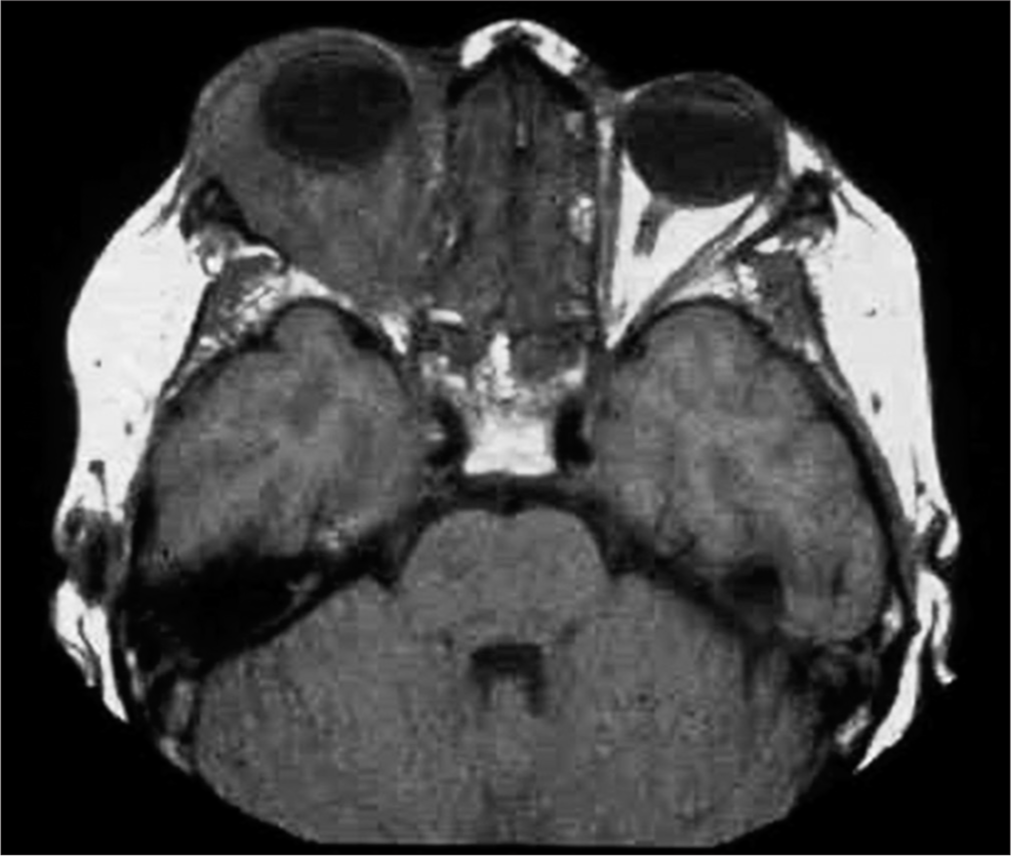
- Granulomatous retro-orbital pseudotumor behind the eye, restricting eye movement and causing diplopia as well as compressing the optic nerve, which untreated can lead to blindness.
Microscopic polyangiitis
The presence of constitutional symptoms of systemic vasculitis is observed at the same frequency in those with MPA as for those with GPA. MPA shares similar organ-specific manifestations with GPA but has less ENT and ocular involvement. There is a higher frequency of renal involvement, with some studies reporting over 90% of patients with MPA having renal disease, compared to around 75% of GPA patients.3 Lower respiratory manifestations are also similar, with alveolar hemorrhage occurring in just under 50%; however, patients with MPA can present (in up to 15% of cases) with isolated pulmonary fibrosis, which is not observed in those with GPA [Figure 4].39
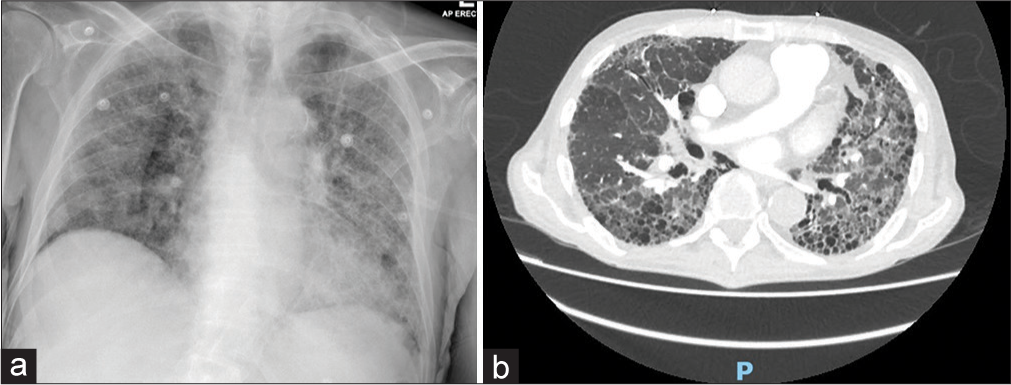
- Pulmonary fibrosis in a patient with microscopic polyangiitis on (a) plain chest X-ray and (b) CT scan.
Renal limited vasculitis
Vasculitis is limited to the kidneys. Patients do not typically demonstrate the systemic features of the disease, as with GPA and MPA. The severity of renal involvement can range from mild injury to severe rapidly progressive GN (RPGN). At its most severe, renal biopsy will reveal a pauci-immune necrotizing crescentic GN [Figure 2a]. RLV is most often associated with p-ANCA/MPO-ANCA in the sera.40
| Clinical features | GPA | MPA |
|---|---|---|
| Constitutional symptoms (fever, weight loss, myalgia, | ++++ | ++++ |
| Ear (hearing loss, otitis media) | ++ | + |
| Nose (nasal crusting, congestion, septal defects) | +++ | + |
| Throat (chronic rhinitis, sinusitis, laryngitis) | + | +/- |
| ENT (endobronchial involvement/inflamed ear or nose cartilage/hoarse voice stridor/saddle nose deformity | ++ | +/- |
| Eyes (pseudotumor, episcleritis, keratitis, conjunctivitis, optic neuritis) | + | +/- |
| Respiratory (SOB, cough, hemoptysis) | +++ | ++ |
| Lower respiratory tract (lung nodules) | ++ | + |
| Pulmonary hemorrhage | + | + |
| Pulmonary fibrosis | - | ++ |
| Renal (hematuria, proteinuria, RPGN) | +++ | ++++ |
| Skin (purpuric/petechial skin rash, ulcers, painful lesions) | + | + |
| PNS (mononeuritis multiplex) | + | + |
| Joints (arthralgia) | +++ | ++ |
| MSK (myalgia, weakness, tenderness) | ++ | ++ |
| GI (abdominal pain, diarrhea) | + | + |
| Laboratory features | ||
| Creatinine >300 | + | +++ |
| Hemoglobin <10 | ++ | +++ |
| Platelets >500 | +++ | ++ |
| Proteinuria | +++ | ++++ |
| Hematuria | +++ | ++++ |
| cANCA | ++++ | +/- |
| Anti-priteinase 3 | ++++ | +/- |
| p-ANCA | + | ++++ |
| Anti-myeloperoxidase | + | ++++ |
| Biopsy findings | ||
| Pauci-immune glomerulonephritis | +++ | +++ |
| Granuloma/giant cells | ++ | - |
| Leuk toclastic vasculitis | + | - |
ENT: Ear, nose, throat, MSK: Musuloskeletal, ANCA: Antineutrohil cytoplasmic antibodies
Diagnosis
Although several classification criteria have been established for AAV, including CHCC 2012 and the recently published 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria, these are designed to help separate patients with similar features into groups to aid recruitment into studies and provide standardization across studies.1,37,38,42 No validated diagnostic criteria exist for AAV. Diagnosis of GPA, MPA, and RLV is based on clinical presentation, aided by the presence of ANCA-positive sera and characteristic fibrinoid necrosis with small vessel inflammatory changes (with or without granuloma) on nasal, lung, kidney, or skin biopsies. As previously mentioned, up to 20% of patients (depending on the presenting phenotype) may be ANCA negative and so, a high degree of suspicion is required to make a diagnosis, particularly early in the disease process.
A nasal biopsy of the inflamed or ulcerated epithelium can reveal necrotizing granulomatous inflammation of the nasal cavity in GPA. However, it frequently only shows chronic inflammatory changes. Biopsy of the upper airways and lungs can reveal characteristic multinucleated giant cells or neutrophilic capillaritis, respectively. Furthermore, lung biopsy may show evidence of alveolar hemorrhage although these are less frequently carried out. Kidney involvement in AAV can vary in severity and show both acute and chronic involvement. Typical features of involvement on light microscopy include segmental fibrinoid necrosis in glomeruli, extracapillary proliferation leading to crescent formation [Figure 2a], which starts as cellular and develops into fibrous crescents. Small vessel extraglomerular vasculitis may be observed depending on the sampling, with fibrinoid necrosis of vessel walls, neutrophil infiltration, and endothelial change [Figure 2b]. As with lung and airway biopsies, granuloma with multinucleated giant cells may be rarely found. Immunofluorescence will show “pauci-immunity,” which refers to the negative/reduced staining for immunoglobulins and complement components.43 In 2010, a classification system for renal histology in AAV was developed and used to prognosticate renal outcomes at 1 and 5 years (see later prognosis section).44
Disease assessment and activity monitoring is an important part of diagnosis and subsequent management of AAV. Several assessment tools have been developed that can be used to assess disease activity, damage from disease, and patient function or quality of life (QoL). Commonly used tools include the Birmingham Vasculitis Activity Score (BVAS), the Vasculitis Damage Index (VDI), and the Health Assessment Questionnaire (HAQ), and short-form 36 (SF-36), both of which assess the function and QoL.45-48 Although not necessarily used in day-to-day clinical practice, as with classification criteria, these tools are useful in clinical trial settings to quantify responses to interventions.
Management
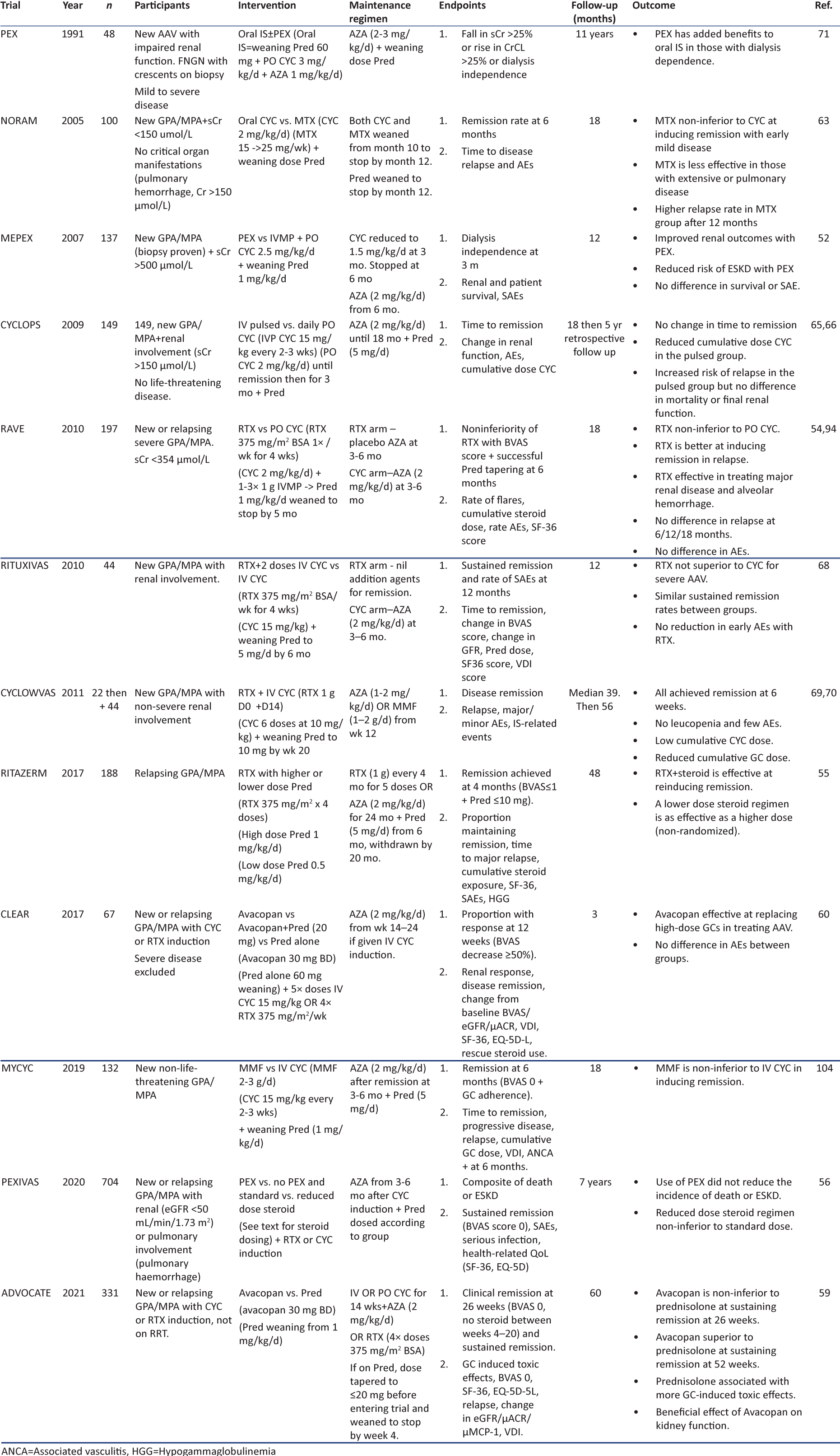
The treatment of AAV is largely the same, regardless of the subtype, and involves the use of immunosuppressive (IS) therapy. There remains a debate as to whether different strategies should be used in PR3- or MPO-AAV beyond managing the risk of relapse; however, to date, no trials have specifically separated the two conditions (although the WGET trial did only enroll GPA patients, the majority of which had PR3-ANCA). The main goals of management are to firstly induce remission of the active disease process and secondly maintain this remission in the long term and prevent relapse of disease. Significant progress has been made in the management of AAV since the first use of IS. The key trials that have contributed to this progress and that dictate the way in which AAV is managed today have been summarized in Tables 2 and 3.
Induction
The goal of induction therapy is to achieve early remission. This remission may be complete (i.e., there are no persistent clinical signs of active inflammatory disease), partial (there are some persistent clinical signs of active disease), or refractory (remission is not achieved). Complicating this process is the exact definition of remission as it is apparent that many immunological processes continue to be active despite clinical quiescence. The type of induction therapy given is dictated by the severity of the disease and extent of organ involvement, with severe disease being classified as being organ- or life-threatening. Induction therapy has traditionally involved the use of glucocorticoids (GCs) and IS agents with or without plasma exchange (PEX). Before starting the IS therapy, all patients should be screened for underlying infections, including human immunodeficiency virus (HIV), tuberculosis (Tb), and hepatitis B/C.49 Clinicians should also be wary of mimics of AAV, for example, patients with undiagnosed infective endocarditis (IE) can present with all features of an acute AAV and ANCA positivity.50 Therapy should be commenced as soon as a diagnosis of AAV seems most probable and once safe to do so.
Glucocorticoids
Glucocorticoids (GCs) have formed the pillar of treatment of AAV for many decades. For those presenting with severe disease (RPGN, pulmonary hemorrhage), pulsed intravenous methylprednisolone (IVMP) (3 doses 0.5–1 g over 3 days) is often administered because it was first shown to be effective at reversing the disease in the 1980s.51 However, the MEPEX trial investigated the role of PEX instead of IVMP in achieving renal recovery in those presenting with severe AAV and sCr > 500 µmol/L; it found PEX to be associated with an increased rate of renal recovery compared with IVMP.52 Retrospective analysis of outcomes of patients treated with both PEX and IVMP suggests no benefit from additional IVMP but increased adverse events, specifically infections and diabetes.53 The role of IVMP in severe disease, therefore, remains controversial with no solid trial-based evidence to demonstrate beneficial outcomes versus adverse effects. However, it continues to be routinely used in clinical practice. For those with less severe disease, IVMP can be omitted.
The high IV dose of GC has historically been followed by, and increasingly replaced by, a gradually weaning dose of oral (PO) GCs, typically prednisolone (or equivalent) starting at 1 mg/kg/d (max. 60–80 mg/d). The side effect profile associated with GC use is well established and includes an increased risk of infection, diabetes, hypertension, and cardiovascular disease (CVD). The morbidity associated with GC use has resulted in many looking for ways in which to reduce cumulative GC dosing.
The RAVE trial aimed to withdraw steroids completely at 6 months, with rituximab (RTX) versus PO cyclophosphamide (CYC) induction, and achieved remission in 64% and 53% of patients, respectively.54 RITAZERM compared RTX induction after relapse of AAV with higher (1 mg/kg/d) and lower (0.5 mg/kg/d) dose steroid regimens and found both to be effective in inducing remission, although steroid dosing groups were non-randomized.55 The PEXIVAS trial also aimed to assess the role of reduced-dose steroids, comparing induction in severe AAV with or without PEX in the presence of high and low-dose steroids. All patients received 1–3 g IVMP, followed by oral GC at 1 mg/kg/d for 7 days. The higher dose group continued at this dose for a further 7 days and then tapered to 12.5–20 mg/d at 3 months and 5 mg/d at 6 months. The lower dose group was weaned to 50% of the starting dose after 7 days and then tapered to 6–10 mg/d at 3 months and 5 mg/d at 6 months. The lower dose group received 60% less cumulative steroid dose than the higher dose group and demonstrated non-inferiority compared to higher GC dose with respect to the primary outcome of death and ESKD. In addition, the reduced steroid group had fewer serious infections at 1 year.56 More recently, the LoVas study compared the standard 1 mg/kg/d prednisolone induction with a 0.5 mg/kg/d dose, without the addition of pulsed methylprednisolone in Japanese patients with less severe AAV. The trial showed equal remission induction rates in the two arms and significantly fewer infections in the lower-dose arm.57 Taken together, these data confirm that lower dose induction GC can be used in AAV with fewer adverse effects and equal efficacy.
More recently, the ADVOCATE trial aimed to determine if PO GC therapy could be largely replaced. This randomized, placebo-controlled trial introduced avacopan, a C5a receptor inhibitor, as a potential alternative to GC therapy. In a double-placebo design trial, PO avacopan (30 mg twice daily) or placebo was administered alongside a tapering dose of PO GC or placebo. Those with organ failure (kidney and lung) were excluded. A small dose of GC was allowed in both groups of patients, including up to 3 g of IVMP for severe AAV (87.3% of the avacopan group received some form of GC). With earlier phase 2 studies (CLEAR and CLASSIC trials) demonstrating tolerability and efficacy of avacopan as an add-on to the standard of treatment and when used alone in a small number of patients, ADVOCATE went on to demonstrated non-inferiority of avacopan to GC at achieving remission after 26 weeks.58-60 Serious adverse events (SAEs) were similar in both groups, but there were fewer GC-related adverse events in the avacopan arm. Avacopan has now received the National Institute for Health and Care Excellence (NICE) and Food and Drug Administration (FDA) approval in the UK and USA, respectively, allowing for its use as an alternative to GC in the induction of remission of GPA and MPA.
Immunosuppressive agents
Cyclophosphamide (CYC) is an alkylating immunosuppressive agent that prevents B and T cell division by inhibiting nuclear DNA replication.61 It was first shown to be effective in the management of AAV in the 1970s.62 CYC was subsequently offered as a daily PO regimen to patients presenting with AAV. However, as with GC, the side effect profile and toxic effects of this medication led to trials to determine ways in which to limit cumulative dosing. Side effects include an increased risk of infections, bone marrow suppression, hemorrhagic cystitis, and an increased risk of solid and hematological malignancies, most notably transitional cell carcinoma of the bladder. The NORAM trial was the first to assess the role of alternative IS therapy in non-severe AAV. This trial compared weekly PO methotrexate (MTX) to daily PO CYC and found MTX to be non-inferior at inducing remission.63 Similarly, the MYCYC trial compared mycophenolate mofetil (MMF) to intravenous (IV) CYC in inducing remission in non-life threatening AAV and showed MMF to be similarly effective to IV CYC.64 Although both NORAM and MYCYC show similar rates of remission with MTX and MMF compared to CYC at 6 months, respectively, they are both associated with increased rates of relapse beyond this. Their use as induction agents should therefore be carefully considered.
CYCLOPS aimed to determine if a reduced cumulative dose of CYC could be effective in inducing remission. It compared daily PO CYC to IV pulsed CYC (3 doses of 15 mg/kg, 1 dose every 2 weeks, then pulses every 3 weeks until remission, then for a further 3 months). No change in time to remission was observed, with a reduced cumulative CYC dose in the IV pulsed group. Although there was an increase in relapse at 1 year in the pulsed group, there was no difference in long-term mortality or final renal function.65,66 Importantly, since using such a regime, fewer cases of hemorrhagic cystitis and bladder malignancy have been reported.67
Rituximab (RTX) is an anti-CD20 monoclonal antibody (mAb) that selectively depletes B cell populations. It was first compared to PO and IV pulsed CYC in the RAVE and RITUXIVAS trials, respectively.54,68 The RAVE trial demonstrated non-inferiority of RTX (375 mg per square meter of body surface area per week for 4 weeks) in inducing remission in severe AAV but excluded those with sCr > 354 µmol/L. No difference in relapse rates or adverse events was observed after 18 months. RITUXIVAS combined RTX (375 mg per square meter of body-surface area per week for 4 weeks) with two IV doses of CYC and compared it to IV CYC alone, demonstrating sustained remission in 76% and 82%, respectively, with no difference in AEs between groups. RITUXIVAS had the added benefit of including patients with severe renal disease, including those on RRT, demonstrating a similar improvement in eGFR between groups.68
Further non-randomized cohort studies of combination therapies have been carried out including CycLowVas, a two-center study, which used RTX (2× 1 g doses 2 weeks apart) and low-dose IV CYC (6× 500 mg doses, each 2 weeks apart) in 66 patients with long-term follow-up. They were able to achieve remission rates of 94% at 6 months, with patient and renal survival of 84 and 95%, respectively, at 5 years. Furthermore, relapse rates in this cohort were surprisingly low, with 70% remaining relapse-free at 5 years. Importantly, this combination therapy allowed for a significant reduction in cumulative GC dosing, limiting dosing to just 1 or 2 weeks.69,70 This, alongside promising results from the RTX arm of RITUXIVAS, with remission rates of 76%, has spurred the wider use of combination induction therapy, which needs to be tested in a randomized trial.
Plasma exchange
Plasma exchange (PEX) involves the separation of plasma from whole blood and the resultant rapid removal of circulating immunoglobulins, including ANCA. PEX was introduced in the 1970s and was subsequently shown to produce favorable outcomes in patients with AAV, particularly in those with dialysis-dependent renal failure and pulmonary hemorrhage.71 As previously mentioned, the MEPEX trial investigated the role of PEX versus IVMP in 137 patients with AAV and severe renal impairment (sCr > 500 umol/L). This trial was able to demonstrate the beneficial role of PEX over IVMP in reducing the risk of ESKD, although it showed no difference in survival or SAEs at 12 months.52 The largest trial carried out to determine the potential beneficial role of PEX in severe AAV is the more recent PEXIVAS trial, which included 704 patients. This 2-by-2 factorial design trial aimed to establish if PEX versus no PEX in the presence of high- and low-dose steroid regimens offered more favorable outcomes with respect to a composite primary endpoint of death and ESKD. This trial was able to demonstrate that PEX did not reduce the risk of all-cause mortality or ESKD after long-term follow-up (up to 7 years). Furthermore, subgroup analysis of the 190 patients presenting with pulmonary hemorrhage showed no added benefit in this group either.56 However, a recent meta-analysis of PEX trials in AAV has shown a significant advantage for using PEX in more severe forms of kidney involvement. An increased rate of developing independent renal function at 1 year was observed; however, it again confirmed no impact on the overall 1-year mortality. The risk and benefit analysis incorporates the increased risk of infection countering the potential benefit of improved kidney function. In patients with a sCr > 500 µmol/l at presentation, there was a clear benefit of PEX, whereas those with relatively preserved renal function benefitted less compared to the risks of treatment. Those with kidney function in between these extremes needed to have careful consideration of the relative benefits.72
Overall, one must be cautious in balancing the risks and benefits of PEX in those presenting with severe AAV. Patient selection may be the key. Additionally, there is evidence to suggest that the use of PEX may benefit patients with active renal inflammation without significant scarring.73
Maintenance
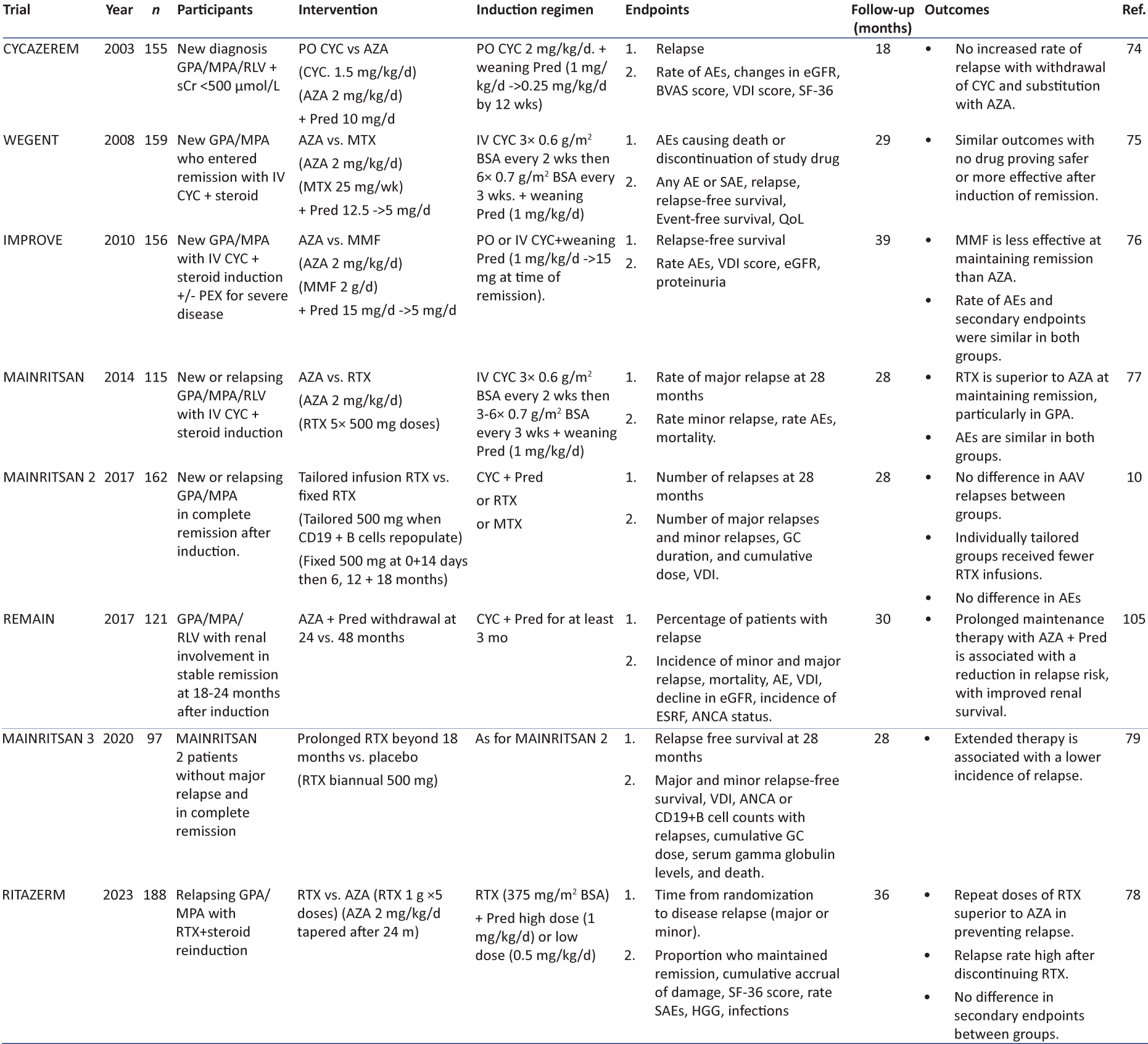
The aim of maintenance therapy is to preserve the state of remission in the long term and avoid disease relapses, thus avoiding further cycles of organ damage. As with induction therapy, several trials have been carried out to determine not only the most effective maintenance regimen, and its duration, but also that which is associated with the fewest side effects [summarized in Table 3. Maintenance regimens have typically involved the use of low-dose GCs and a PO IS agent continued for at least 18–24 months before considering withdrawal. Some newer trial protocols have tried to tail off GC completely at earlier stages, whereas other studies have focused on the ideal duration of maintenance therapy. The REMAIN trial compared extending maintenance therapy beyond 24 months to 48 months. There was a lower relapse rate in the extended maintenance group versus the withdrawal group, 22% versus 62.7%, respectively, but more side effects in those continuing treatment for longer. Other studies examining the ideal duration of maintenance have provided variable results, and the total number of patients studied has been small.
Azathioprine (AZA) has been the PO IS of choice for maintenance therapy for many years as a result of data from several trials. The CYCAZERM trial assessed the role of AZA versus PO CYC in maintaining remission after PO CYC and GC induction and found both to be associated with similar relapse rates. Although AEs were similar in both groups (11% in AZA, 10% in CYC), the use of AZA over PO CYC allows for reduced cumulative doses of CYC, therefore limiting the associated complications of high CYC exposure.74 Later trials aimed to determine if alternative PO IS agents would be as effective as AZA at maintaining remission but with fewer AEs. WEGENT compared AZA to MTX after IV CYC and GC induction, finding similar rates of relapse in both, with no drug proving safer or more effective.75 The IMPROVE trial comparing MMF to AZA found MMF to be inferior at maintaining remission, with similar AEs in both groups.76
Despite these trials demonstrating the efficacy of AZA and MTX in maintaining remission, relapse rates still remain high (30–50%). Several trials went on to determine if IV maintenance IS could be superior. The MAINRITSAN 1, followed by the RITAZERM, trial assessed if maintenance RTX improved relapse rates compared to AZA. The MAINRITSAN 1 studied new AAV patients administered CYC + GC induction, then RTX maintenance (500 mg every 6 months), and RITAZERM observed relapsed AAV patients re-induced with RTX and GC then administered RTX maintenance (1 g every 4 months). Both trials found RTX to be associated with fewer relapses after 28 and 36 months, respectively.77,78 MAINRITSAN 2 went on to determine if RTX dosing tailored to B-cell population counts was as effective as scheduled RTX dosing. They found no difference in relapse rates, with the tailored arm receiving fewer infusions overall. In both MAINRITSAN 1 and 2, AEs and SAEs were similar across groups. Finally, MAINRITSAN 3 aimed to determine if biannual RTX (500 mg every 6 months) beyond 18 months, versus placebo, would successfully prevent relapse in patients who were in complete remission and who had completed MAINRITSAN 2 without any major relapse. At 28 months, they were able to achieve relapse-free survival in 96% and 74% in the biannual RTX and placebo groups, respectively, with an absolute difference of 22% and a hazards ratio of 7.5.79
Alongside PO and IV maintenance IS, GC has been used in the prevention of relapse. During induction therapy, GC doses are gradually weaned to <10 mg/day over a 3–6 month period. Many will continue on low-dose steroids for a further 12–18 months, before trying to gradually withdraw therapy. As with induction regimens, more recent trials have looked into ways to limit long-term GC exposure, and their associated side effects. As mentioned earlier the ADVOCATE trial, in addition to observing induction of remission, also compared the role of avacopan to prednisolone in maintaining remission. Patients were randomly allocated to receive avacopan (30 mg twice daily) or a tapering schedule of prednisolone, which stopped at month 6, following CYC then AZA (2 mg/kg/day), or RTX induction. Remission at 26 and 52 weeks in the avacopan and prednisolone groups was 72.3% and 65.7%, and 70.1% and 54.9%, respectively. Avacopan was therefore deemed non-inferior to prednisolone at 26 weeks and superior at 52 weeks.59 However, it must be noted that between months 6 and 12, the groups were not truly matched as one had avacopan therapy and the other no steroids at all, making the claims of superiority somewhat overstated. Having now received NICE and FDA approval for maintenance, as well as induction therapy, we are likely to observe an increase in the use of avacopan and an associated reduction in long-term GC use for the management of AAV.
Additional therapy
For those who receive GC therapy, it is important for any clinician to ensure that adequate precautions are taken to limit long-term sequelae of use. Patients should routinely be commenced on proton pump inhibitors or H2 receptor antagonists alongside vitamin D and calcium supplements for gastric and bone protection, respectively. With the increased risk of developing diabetes and hypercholesterolemia, HbA1c and cholesterol levels should be checked at regular intervals.
IS therapy is associated with an increased risk of developing opportunistic infections, such as Pneumocystis jirovecii, and so patients should be commenced on co-trimoxazole or allergic alternatives such as dapsone, atovaquone, or pentamidine to reduce this risk. Similarly, Candida infections can develop; thus, prophylactic fluconazole or nystatin is often administered. In at-risk groups, with a higher likelihood of previous exposure to Tb, prophylactic isoniazid may also be commenced. Patients should receive routine vaccinations with pneumococcal, influenza, COVID-19, and hepatitis B vaccines and should be advised to avoid live vaccines.
In the long term, patients are known to be at increased risk of CVD and malignancy; thus, it is vital that clinicians monitor for these and take appropriate action where necessary.80,81
Relapse and biomarkers of disease
Thanks to advancements in therapy, AAV has evolved from a life-limiting to a chronic disease state. With increased survival comes the increased risk of relapse, as such, relapse is common with rates ranging from 30 to 50% over 5 years.82,83 As demonstrated by the CYCLOPS and MAINRTISAN 3 trials, increased and prolonged IS therapy are known to be associated with reduced relapse rates; however, it is important to find a balance between the risk of organ damage caused by relapse and the consequences of excessive long-term IS therapy. Identifying those at the greatest risk of relapse who may need more therapy and those who are less likely to relapse is therefore a critical area of ongoing investigation to allow customized therapies for patients.
Several risk factors for disease relapse have been identified with PR3-ANCA positivity, low sCr, and cardiovascular system involvement being the risk factors with the strongest association.83 In addition, the presence of ENT system involvement, persistence, or increase in ANCA titers after treatment, discontinuation of short duration IS, lower cumulative dose of CYC, discontinuation of GC, B-cell reconstitution post-RTX, and chronic nasal carriage of Staphylococcus aureus are associated with an increased risk of relapse.24,82,84,85 Despite these known risk factors, it remains difficult to not only predict which patients will relapse but also know when to diagnose a relapse. Biomarkers, such as ANCA titers and B-cell population reconstitution can be a helpful guide but are not always concordant with clinical activity or occurrence of relapse. Many have made efforts to identify more sensitive biomarkers for disease activity. Possible candidate biomarkers have been identified, including urinary monocyte chemoattractant protein-1 (MCP-1), a soluble cluster of differentiation 163 (CD163), and urinary T cell populations; however, many of these remain experimental and are yet to be validated for use in routine clinical practice.86-89 An ongoing study, data-driven identification of AAV relapse in real-world trials, aims to create an algorithm that will allow for a more accurate diagnosis of relapse outside of clinical trials.90
For those who do experience a relapse of disease, management involves administering further induction agents. As a rule, one would tend to opt for an IS agent not previously given that is, if previously given CYC induction, then RTX would be given and vice versa. For those with particularly refractory disease, IV immunoglobulin (IVIG) may be given at a dose of 0.4 g/kg/day for 5 days (max. dose 2 g/kg). IVIG was shown to be associated with reduced disease activity, although this effect was not maintained beyond 3 months.91 The ALEVIATE trial assessed the efficacy and safety of alemtuzumab, a lymphocyte-depleting anti-CD52 antibody, in refractory and relapsing AAV. Remission was achieved in 70% with alemtuzumab at 6 months and maintained in a third of patients at 12 months.92 The safety profile was deemed acceptable however, without data from an RCT, Alemtuzumab is limited in its use for refractory or relapsing AAV.
Future therapies
Therapies continue to be developed and tested in the management of AAV. Abatacept is a CTLA4-immunoglobulin that blocks co-stimulatory signals required for T-cell activation. An open-label trial was carried out to assess the efficacy and safety of abatacept in patients with non-severe relapsing GPA. Abatacept achieved remission in 80% of patients, with 73% able to wean from GC. However, the sample size was small and patients with severe GPA were excluded, making it difficult to draw conclusions from these results.93
The COMBIVAS trial, currently in follow-up, observed the effect of RTX alone versus a combination of RTX and belimumab, a human monoclonal antibody that inhibits the activity of B-cell activating factor (BAFF) resulting in B cell apoptotic death, in PR3-ANCA vasculitis.94 The idea behind this combination therapy is to enhance B cell targeting and depletion, thus reducing relapse rates. Results are due to be published in 2023.95
ObiVas is a randomized controlled trial, which remains open to recruitment, observing the induction of remission with RTX versus obinutuzumab. Obinutuzumab is a type II mAb to CD20, which has a higher affinity for CD20 compared to RTX and has demonstrated superior B-cell depletion in previous clinical trials. The hope is that with improved B-cell depletion, relapse rates will fall with obinutuzumab.96
Currently, the treatment for AAV is largely guided by the severity of AAV, rather than ANCA or clinical subtype. As previously discussed, different subtypes of AAV differ not only in prevalence amongst different populations globally but also in terms of organ involvement and relapse rates. Further to this, dosing and drug regimens are developed for specific clinical trial populations and often do not factor in age, sex, or genetic differences amongst those suffering from AAV on a global scale. A consideration for future therapies is to determine if therapies for AAV can be more individualized, and therefore optimized, for individual AAV subtypes and patients.
Other considerations
AAV is a multisystem disease that can have long-lasting consequences, the role of a multidisciplinary team (MDT) is therefore essential in managing patients to ensure optimal care. In addition to the MDT, successful management of patients requires appropriate patient education and engagement with their condition. Although the ultimate aim of treatment is to induce and maintain remission of disease, it is important to always consider what impact treatments have on different patients and their QoL, and how this can be improved in the long term. With appropriate education, patients’ expectations and understanding of their condition can be managed, which in itself can contribute to improved QoL.
Prognosis
The prognosis of patients with AAV has significantly improved since the introduction of GC and IS in their management. Studies, however, continue to demonstrate that patients with AAV are at increased risk of death, at all ages, compared to the general population. Infections, CVD, and malignancy account for the main causes of death.97 It is perhaps unsurprising that patients presenting with significant organ failure, including significant renal impairment and diffuse pulmonary hemorrhage, have an increased risk of dying.98 Similarly, late remission, refractory disease, and early relapse are associated with progressive organ damage and therefore worse prognosis. Older age at the time of diagnosis, male sex, and low platelet count are also all negative prognostic factors.97,99
Renal involvement is common in AAV and development of ESKD is associated with significant long-term implications, including increased risk of CVD and mortality.100 Approximately 20% of patients with nephritis will develop ESKD, whereas those that have significant kidney involvement at presentation (eGFR < 50) will have a 50% risk of dying or developing ESKD within 5 years.101 Renal biopsy is the gold standard method for diagnosing renal involvement in AAV. The Berden classification system was developed to help categorize histological findings on renal biopsy to prognosticate renal outcomes at the time of diagnosis. Four categories were defined; focal, crescentic, mixed, and sclerotic. Table 4 summarizes the categories and their associated predicted 1- and 5-year renal survival rates.44 Similarly, Brix et al. developed a renal risk score to predict the risk of ESKD at 36 months based on renal biopsy findings at the time of diagnosis. This score observes the number of normal glomeruli, the degree of interstitial fibrosis and tubular atrophy and includes the presenting eGFR.102 The Brix score was able to separate the cohort into low, intermediate, and high risk for ESKD. It is important to recognize that both the Brix and Berden scoring systems used retrospective data and patient inclusion was limited to those with particular follow-ups, meaning important data may have been omitted. Patients enrolled in these studies had been treated with standard therapies. As such, these scores should not be used to decide on whether to treat or not, but are helpful in managing expectations for disease outcomes. Finally, the use of new agents may alter the potential for renal recovery, exemplified by the findings that avacopan use was associated with a greater improvement in GFR compared to standard steroids in the Advocate study, most noticeably in those with the worst starting GFR.103
 |
 |
| Class | Criteria | 1-year renal survival (%) | 5-year renal survival (%) |
|---|---|---|---|
| Focal | ≥50% glomeruli are normal | 93 | 93 |
| Crescentic | Cellular crescents in ≥50% glomeruli | 84 | 76 |
| Sclerotic | ≥50% glomeruli globally sclerotic | 50 | 50 |
| Mixed | <50% normal/crescentic/globally. Sclerotic glomeruli | 69 | 61 |
Conclusion
AAV are rare conditions with broad clinical presentations and significant long-term consequences. Renal disease remains one of the most important predictors of outcome. The management of these conditions has evolved significantly over recent decades, with patient survival increasing in line with this. However, there remains room for improvement in therapy, with future efforts focusing on reducing relapse rates, reducing toxic side effects, and long-term consequences of IS, and improving individualization of care.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
T Roper is supported by a Clevland Clinic PhD studentship.
References
- 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- [CrossRef] [PubMed] [Google Scholar]
- A historical study of American patients with anti-neutrophil cytoplasmic antibody negative pauci-immune glomerulonephritis. Clin Rheumatol. 2016;35:953-60.
- [CrossRef] [PubMed] [Google Scholar]
- Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: A cluster analysis. Ann Rheum Dis. 2013;72:1003-10.
- [CrossRef] [PubMed] [Google Scholar]
- Giant-cell granuloma of thfe respiratory tract (wegener's granulomatosis) BMJ. 1958;2:265-70.
- [CrossRef] [PubMed] [Google Scholar]
- Overall survival, renal survival and relapse in patients with microscopic polyangiitis: A systematic review of current evidence. Rheumatology (Oxford). 2011;50:1414-23.
- [CrossRef] [PubMed] [Google Scholar]
- Systemic vasculitis in the 1980s-Is there an increasing incidence of Wegener's granulomatosis and microscopic polyarteritis? J R Coll Physicians Lond. 1990;24:284-8.
- [Google Scholar]
- An update on the epidemiology of ANCA-associated vasculitis. Rheumatology (Oxford). 2020;59(Suppl 3):iii42-50.
- [CrossRef] [PubMed] [Google Scholar]
- Geoepidemiology of systemic vasculitis: Comparison of the incidence in two regions of Europe. Ann Rheum Dis. 2001;60:170-2.
- [CrossRef] [PubMed] [Google Scholar]
- Global ethnic and geographic differences in the clinical presentations of anti-neutrophil cytoplasm antibody-associated vasculitis. Rheumatology (Oxford). 2017;56:1962-9.
- [CrossRef] [PubMed] [Google Scholar]
- Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the U.K. Rheumatology (Oxford). 2011;50:1916-20.
- [CrossRef] [PubMed] [Google Scholar]
- Wegener's granulomatosis in New Zealand: Evidence for a latitude-dependent incidence gradient. Intern Med J. 2007;37:242-6.
- [CrossRef] [PubMed] [Google Scholar]
- Incidence of ANCA-associated vasculitis in a UK mixed ethnicity population. Rheumatology (Oxford). 2016;55:1656-63.
- [CrossRef] [PubMed] [Google Scholar]
- Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrol Dial Transplant. 2015;30(Suppl 1):i14-22.
- [CrossRef] [PubMed] [Google Scholar]
- A cohort study to investigate sex-specific differences in ANCA-associated glomerulonephritis outcomes. Sci Rep. 2021;11:13080.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology of systemic vasculitis: A ten-year study in the United Kingdom. Arthritis Rheum. 2000;43:414-9.
- [CrossRef] [PubMed] [Google Scholar]
- Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955-63.
- [CrossRef] [PubMed] [Google Scholar]
- Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood. 2005;106:2050-8.
- [CrossRef] [PubMed] [Google Scholar]
- Colocalization of ANCA-antigens and fibrinoid necrosis in ANCA-associated vasculitis. Kidney Int. 2001;60:2025-30.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology of vasculitis in Europe. Ann Rheum Dis. 2001;60:1156-7.
- [CrossRef] [PubMed] [Google Scholar]
- Environmental risk factors associated with ANCA associated vasculitis: A systematic mapping review. Autoimmun Rev. 2020;19:102660.
- [CrossRef] [PubMed] [Google Scholar]
- Antineutrophil cytoplasmic antibody-associated vasculitides: Could geographic patterns be explained by ambient ultraviolet radiation? Arthritis Rheum. 2009;61:1417-24.
- [CrossRef] [PubMed] [Google Scholar]
- The association between silica exposure and development of ANCA-associated vasculitis: Systematic review and meta-analysis. Autoimmun Rev. 2013;12:1129-35.
- [CrossRef] [PubMed] [Google Scholar]
- The influence of the Great East Japan earthquake on microscopic polyangiitis: A retrospective observational study. PLoS One. 2017;12:e0177482.
- [CrossRef] [PubMed] [Google Scholar]
- Association of chronic nasal carriage of staphylococcus aureus and higher relapse rates in wegener granulomatosis. Ann Intern Me. 1994;120:12-7.
- [CrossRef] [PubMed] [Google Scholar]
- Using a network-based analysis approach to investigate the involvement of S. aureus in the pathogenesis of granulomatosis with polyangiitis. Int J Mol Sci. 2023;24:1822.
- [CrossRef] [PubMed] [Google Scholar]
- Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody associated vasculitis: The cause is hidden, but the result is known. Clin J Am Soc Nephrol. 2008;3:237-52.
- [CrossRef] [PubMed] [Google Scholar]
- Vasculitis and antineutrophil cytoplasmic autoantibodies associated with propylthiouracil therapy. Lancet. 1993;11:651-2.
- [CrossRef] [PubMed] [Google Scholar]
- Trojan horses: Drug culprits associated with antineutrophil cytoplasmic autoantibody (ANCA) vasculitis. Curr Opin Rheumatol. 2014;26:42-9.
- [CrossRef] [PubMed] [Google Scholar]
- Immune checkpoint inhibitors as potential triggers for ANCA vasculitis. RMD Open. 2022;8:e002500.
- [CrossRef] [PubMed] [Google Scholar]
- Vasculitis associated with immune checkpoint inhibitors-a systematic review. Clin Rheumatol. 2018;37:2579-84.
- [CrossRef] [PubMed] [Google Scholar]
- De novo and relapsing necrotizing vasculitis after COVID-19 vaccination. Clin Kidney J. 2022;15:560-3.
- [CrossRef] [PubMed] [Google Scholar]
- Vasculitis following influenza vaccination: A review of the literature. Curr Rheumatol Rev. 2017;13:188-96.
- [CrossRef] [PubMed] [Google Scholar]
- Cocaine-induced granulomatosis with polyangiitis-an under-recognized condition. Rheumatol Adv Pract. 2023;7:rkad027.
- [CrossRef] [PubMed] [Google Scholar]
- Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. 2012;367:214-23.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Rheumatol. 2017;69:1054-66.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic variants in antineutrophil cytoplasmic antibody-associated vasculitis: A bayesian approach and systematic review. J Clin Med. 2019;8:266.
- [CrossRef] [PubMed] [Google Scholar]
- 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for granulomatosis with polyangiitis. Ann Rheum Dis. 2022;81:315-20.
- [CrossRef] [PubMed] [Google Scholar]
- 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis. Ann Rheum Dis. 2022;81:321-6.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J. 2010;36:116-21.
- [CrossRef] [PubMed] [Google Scholar]
- Classification and characteristics of Japanese patients with antineutrophil cytoplasmic antibody-associated vasculitis in a nationwide, prospective, inception cohort study. Arthritis Res Ther. 2014;16:R101.
- [CrossRef] [PubMed] [Google Scholar]
- Classification of ANCA-associated vasculitis: Differences based on ANCA specificity and clinicopathologic phenotype. Rheumatol Int. 2021;41:1717-28.
- [CrossRef] [PubMed] [Google Scholar]
- Imnmnopathological aspects of systemic vasculitis. Springer Semin Irnmunopathol. 2001;23:253-65.
- [CrossRef] [PubMed] [Google Scholar]
- Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21:1628-36.
- [CrossRef] [PubMed] [Google Scholar]
- Birmingham vasculitis activity score (BVAS) in systemic necrotizing vasculitis. QJM. 1994;87:671-8.
- [Google Scholar]
- Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum. 1997;40:371-80.
- [CrossRef] [PubMed] [Google Scholar]
- The dimensions of health outcomes: The health assessment questionnaire, disability and pain scales. J Rheumatol. 1982;9:789-93.
- [Google Scholar]
- The MOS 36-item short-form health survey (SF-36) I. Conceptual framework and item selection. Med Care. 1992;30:473-83.
- [CrossRef] [PubMed] [Google Scholar]
- Reactivation of hepatitis B virus infection in rheumatic diseases: Risk and management considerations. Ther Adv Musculoskelet Dis. 2020;12:1759720X20912646.
- [CrossRef] [PubMed] [Google Scholar]
- Infective endocarditis with antineutrophil cytoplasmic antibody: Report of 13 cases and literature review. PLoS One. 2014;9:e89777.
- [CrossRef] [PubMed] [Google Scholar]
- Methylprednisolone therapy for acute crescentic rapidly progressive glomerulonephritis. Am J Nephrol. 1989;9:368-75.
- [CrossRef] [PubMed] [Google Scholar]
- Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180-8.
- [CrossRef] [PubMed] [Google Scholar]
- Intravenous pulse methylprednisolone for induction of remission in severe ANCA associated Vasculitis: A multi-center retrospective cohort study. BMC Nephrol. 2019;20:58.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;2010:221-32.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab as therapy to induce remission after relapse in ANCA-associated vasculitis. Ann Rheum Dis. 2020;79:1243-9.
- [CrossRef] [PubMed] [Google Scholar]
- Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med. 2020;382:622-31.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of reduced-dose vs high-dose glucocorticoids added to rituximab on remission induction in ANCA-associated vasculitis: A randomized clinical trial. JAMA. 2021;325:2178-87.
- [CrossRef] [PubMed] [Google Scholar]
- Adjunctive treatment with avacopan, an oral C5a receptor inhibitor, in patients with antineutrophil cytoplasmic antibody-associated vasculitis. ACR Open Rheumatol. 2020;2:662-71.
- [CrossRef] [PubMed] [Google Scholar]
- Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. 2021;384:599-609.
- [CrossRef] [PubMed] [Google Scholar]
- Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. 2017;28:2756-67.
- [CrossRef] [PubMed] [Google Scholar]
- The immunopharmacology of cyclophosphamide. Int J Immunopharmacol. 1979;1:165-71.
- [CrossRef] [PubMed] [Google Scholar]
- Cyclophosphamide therapy in wegener's Granulomatosis. N Engl J Med. 1971;284:938-42.
- [CrossRef] [PubMed] [Google Scholar]
- Randomized trial of cyclophoshamide veresus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005;52:2461-9.
- [CrossRef] [PubMed] [Google Scholar]
- Mycophenolate mofetil versus cyclophosphamide for the induction of remission in nonlife-threatening relapses of antineutrophil cytoplasmic antibody-associated vasculitis. Clin J Am Soc Nephrol. 2019;14:1021-8.
- [CrossRef] [PubMed] [Google Scholar]
- Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: A randomized trial. Ann Intern Me. 2009;150:670-80.
- [CrossRef] [PubMed] [Google Scholar]
- Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: Long-term follow-up. Ann Rheum Dis. 2012;71:955-60.
- [CrossRef] [PubMed] [Google Scholar]
- Incidence and prevention of bladder toxicity from cyclophosphamide in the treatment of rheumatic diseases: A data-driven review. Arthritis Rheum. 2010;62:9-21.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211-20.
- [CrossRef] [PubMed] [Google Scholar]
- Prolonged disease-free remission following rituximab and low-dose cyclophosphamide therapy for renal ANCA-associated vasculitis. Nephrol Dial Transplant. 2011;26:3280-6.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term follow-up of a combined rituximab and cyclophosphamide regimen in renal anti-neutrophil cytoplasm antibody-associated vasculitis. Nephrol Dial Transplant. 2019;34:63-73.
- [CrossRef] [PubMed] [Google Scholar]
- Plasma exchange in focal necrotizing glomerulonephritis without anti-GBM antibodies. Kidney Int. 1991;40:757-63.
- [CrossRef] [PubMed] [Google Scholar]
- The effects of plasma exchange in patients with ANCA-associated vasculitis: An updated systematic review and meta-analysis. BMJ. 2022;376:e064604.
- [CrossRef] [PubMed] [Google Scholar]
- Kidney histopathology can predict kidney function in ANCA-associated vasculitides with acute kidney injury treated with plasma exchanges. J Am Soc Nephrol. 2022;33:628-37.
- [CrossRef] [Google Scholar]
- A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. NEJM. 2003;349:36-44.
- [CrossRef] [PubMed] [Google Scholar]
- Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 2008;359:2790-803.
- [CrossRef] [PubMed] [Google Scholar]
- Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis. A randomized controlled trial. JAMA. 2010;304:2381-8.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371:1771-80.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab versus azathioprine for maintenance of remission for patients with ANCA-associated vasculitis and relapsing disease: An international randomised controlled trial. Ann Rheum Dis. 2023;82:937-44.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term rituximab use to maintain remission of antineutrophil cytoplasmic antibody-associated vasculitis: A randomized trial. Ann Intern Med. 2020;173:179-87.
- [CrossRef] [Google Scholar]
- Malignancies in patients with antineutrophil cytoplasmic antibody-associated vasculitis: A population-based cohort study. J Rheumatol. 2020;47:1229-37.
- [CrossRef] [PubMed] [Google Scholar]
- Chronic inflammatory disorders and risk of type 2 diabetes mellitus, coronary heart disease, and stroke: A population-based cohort study. Circulation. 2014;130:837-44.
- [CrossRef] [PubMed] [Google Scholar]
- Risk factors for relapse of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012;64:542-8.
- [CrossRef] [PubMed] [Google Scholar]
- Predicting relapse in anti-neutrophil cytoplasmic antibody-associated vasculitis: A Systematic review and meta-analysis. Rheumatol Adv Pract. 2021;5:rkab018.
- [CrossRef] [PubMed] [Google Scholar]
- Negative anti-neutrophil cytoplasm antibody at switch to maintenance therapy is associated with a reduced risk of relapse. Arthritis Res Ther. 2017;19:129.
- [CrossRef] [PubMed] [Google Scholar]
- Relapse in anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitis. Kidney Int Rep. 2020;5:7-12.
- [CrossRef] [PubMed] [Google Scholar]
- Urinary monocyte chemoattractant protein-1 (MCP-1) is a marker of active renal vasculitis. Nephrol Dial Transplant. 2004;19:2761-8.
- [CrossRef] [PubMed] [Google Scholar]
- Urinary soluble CD163 in active renal vasculitis. J Am Soc Nephrol. 2016;27:2906-16.
- [CrossRef] [PubMed] [Google Scholar]
- Circulating neutrophil gelatinase-associated lipocalin: A useful biomarker for assessing disease activity of ANCA-associated vasculitis. Rheumatology (Oxford). 2009;48:355-8.
- [CrossRef] [PubMed] [Google Scholar]
- Urinary T cells identify renal antineutrophil cytoplasmic antibody-associated vasculitis and predict prognosis: A proof of concept study. Kidney Int Rep. 2023;8:871-83.
- [CrossRef] [PubMed] [Google Scholar]
- #4553 Real world data-driven identification of anca vasculitis relapse. Nephrol Dial Transplant. 2023;38(Suppl_1) doi: 10.1093/ndt/gfad063c_4553
- [CrossRef] [Google Scholar]
- Intravenous immunoglobulin for ANCA-associated systemic vasculitis with persistent disease activity. QJM. 2000;93:433-9.
- [CrossRef] [PubMed] [Google Scholar]
- Alemtuzumab for refractory primary systemic vasculitis-a randomised controlled dose ranging clinical trial of efficacy and safety (ALEVIATE) Arthritis Res Ther. 2022;24:81.
- [CrossRef] [PubMed] [Google Scholar]
- An open-label trial of abatacept (CTLA4-IG) in non-severe relapsing granulomatosis with polyangiitis (Wegener's) Ann Rheum Dis. 2014;73:1376-9.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab versus cyclophosphamide for ANCA-associated vasculitis with renal involvement. J Am Soc Nephrol. 2015;26:976-85.
- [CrossRef] [PubMed] [Google Scholar]
- A randomised study of rituximab and belimumab sequential therapy in PR3 ANCA-associated vasculitis (COMBIVAS): Design of the study protocol. Trials. 2023;24:180.
- [CrossRef] [PubMed] [Google Scholar]
- Obinutuzumab compared with rituximab for treating ANCA-associated vasculitis. Rheumatology (Oxford). 2022;61:3814-7.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term outcomes and prognostic factors for survival of patients with ANCA-associated vasculitis. Nephrol Dial Transplant. 2023;38:1655-65.
- [CrossRef] [PubMed] [Google Scholar]
- Renal survival in proteinase 3 and myeloperoxidase ANCA-associated systemic vasculitis. Clin J Am Soc Nephrol. 2013;8:1709-17.
- [CrossRef] [PubMed] [Google Scholar]
- Outcome of ANCA-associated renal vasculitis: A 5-year retrospective study. Am J Kidney Dis. 2003;41:776-84.
- [CrossRef] [PubMed] [Google Scholar]
- Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: A systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis. 2008;67:1004-10.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term patient survival in ANCA-associated vasculitis. Ann Rheum Dis. 2011;70:488-94.
- [CrossRef] [PubMed] [Google Scholar]
- Development and validation of a renal risk score in ANCA-associated glomerulonephritis. Kidney Int. 2018;94:1177-88.
- [CrossRef] [PubMed] [Google Scholar]
- Renal recovery for patients with ANCA-associated vasculitis and low eGFR in the Advocate trial of AVACOPAN. Kidney Int Rep. 2023;8:860-70.
- [CrossRef] [PubMed] [Google Scholar]
- Mycophenolate mofetil versus cyclophosphamide for remission induction in ANCA-associated vasculitis: A randomised, non-inferiority trial. Ann Rheum Dis. 2019;78:399-405.
- [CrossRef] [PubMed] [Google Scholar]
- Randomised controlled trial of prolonged treatment in the remission phase of ANCA-associated vasculitis. Ann Rheum Dis. 2017;76:1662-8.
- [CrossRef] [PubMed] [Google Scholar]




