

Translate this page into:
ANCA-Associated Vasculitis with Systemic Thrombotic Microangiopathy: A Review of Literature
Corresponding author: Dr. Raja Ramachandran, Department of Nephrology, Postgraduate Institute of Medical Education and Research, Chandigarh, India. E-mail: drraja_1980@yahoo.co.in
-
Received: ,
Accepted: ,
How to cite this article: Shukla S, Sekar A, Naik S, Rathi M, Sharma A, Nada R, et al. ANCA-Associated Vasculitis with Systemic Thrombotic Microangiopathy: A Review of Literature. Indian J Nephrol. 2024;34:155–61. doi: 10.4103/ijn.ijn_376_22
Abstract
Introduction:
Antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV) rarely coexist with systemic thrombotic microangiopathy (TMA).The TMA can be in the form of either hemolytic uremic syndrome (HUS) or thrombotic thrombocytopenic purpura (TTP). This review explores the clinical characteristics, histopathological findings, treatment options, and outcomes in patients presenting as AAV with coexisting HUS/TTP.
Methods:
We conducted a search on the PubMed database and additional searches from January 1998 to September 2022 using the following terms: “ANCA”, “Antineutrophil cytoplasmic antibody”, “thrombotic thrombocytopenic purpura”, “TTP”, “thrombotic microangiopathy”, “haemolytic uremic syndrome”, and “HUS”. We excluded articles that described renal-limited TMA. Two authors independently reviewed the full texts and extracted all critical data from the included case reports. Finally, we included 15 cases for this review. Hematological remission and kidney recovery in the form of independence from dialysis was assessed.
Results:
The median age of the patients was 61 years and a majority of them were females (66.7%). Myeloperoxidase (MPO)-ANCA positivity (66.67%) was more common than proteinase 3 (PR3)-ANCA positivity (33.33%). All patients had laboratory parameters consistent with systemic TMA (HUS or TTP), and only six (out of 11) cases showed histological features of renal TMA. Ten had crescentic glomerulonephritis, and two had advanced degrees of chronicity in histology. Eighty-six percent of cases had hematological remission, and sixty percent of cases became dialysis-independent after treatment.
Conclusion:
In conclusion, kidney outcome was worse in patients who manifested both AAV and systemic TMA. A paucity of literature regarding this diagnostic quandary calls for avid reporting of such cases.
Keywords
AAV
ANCA
HUS
TMA
TTP

Introduction
Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis (AAV) is characterized by inflammation of predominantly small- and medium-sized blood vessels, resulting in endothelial injury and tissue damage. These are autoimmune diseases due to antibodies to leukocyte proteinase 3 (PR3) or myeloperoxidase (MPO). The three subtypes of AAV are granulomatous polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatous polyangiitis (EGPA). They are defined according to their clinical features. Since the genetic differences, treatment response, and outcomes are specific to the type of ANCA (PR3 or MPO), AAV is better classified as PR3-AAV or MPO-AAV.1 The hallmark of GPA includes upper and lower respiratory tract involvement, pulmonary hemorrhage, granulomatous inflammation, and necrotizing glomerulonephritis. MPA is usually associated with severe renal involvement and some features of GPA without granulomatous inflammation. Additionally, thrombocytosis is a conspicuous feature of AAV.2
On the contrary, systemic thrombotic microangiopathy (TMA) is characterized by microangiopathic hemolytic anemia and thrombocytopenia, with a varying degree of impairment to the kidney, brain, lungs, pancreas, and the heart. Various diseases like systemic lupus erythematosus and cryoglobulinemia are associated with TMA; however, the association of AAV and TMA is a rare occurrence.3,4
Though multiorgan dysfunction is a regular feature of both AAV and systemic TMA, the etiopathogenesis and histology of both the diseases are discrete. Nonetheless, dysregulation of the alternative pathway of the complement cascade is critical to the pathogenesis of both AAV and hemolytic uremic syndrome (HUS). The simultaneous manifestation of both AAV and HUS is rare and exceptional. Although a retrospective review was recently published that described the clinicopathological characteristics of renal-limited TMA in AAV, literature on systemic TMA in AAV has been limited to only a few case reports.5 Thus, we report an imperative retrospective review that describes the baseline characteristics, laboratory findings, treatment, and outcome of patients with AAV and systemic TMA (HUS and thrombotic thrombocytopenic purpura [TTP]).
Materials and Methods
We conducted a search on the PubMed database and additional searches from January 1998 to September 2022 using the following terms: “ANCA”, “Antineutrophil cytoplasmic antibody”, “thrombotic thrombocytopenic purpura”, “TTP”, “thrombotic microangiopathy”, “haemolytic uremic syndrome”, and “HUS”. In addition to this, we selected articles in the English language for review, if their title or abstract suggested AAV with TTP, HUS, or TMA. We excluded reports if they described renal-limited TMA or if the patient developed end-stage kidney disease (ESKD) before manifesting TMA. The title, abstracts, and the full text of the case reports were individually checked by two authors (SS and SN) and considered for inclusion if both agreed.
Statistical analysis
Descriptive statistics were used to detail the baseline characteristics of the patients. We expressed the normally distributed continuous variables as mean ± standard deviation (range), non-normally distributed variables as medians with interquartile ranges (IQR), and categorical data as proportions. All analyses were performed using GraphPad Prism version 9 (San Diego, CA 92108).
Results
Our database search led us to 120 English language publications on PubMed. Details of these studies are shown in Figure 1. Out of 120 publications, we identified 14 cases, including our own case report, and one additional case from web-based search. Thus, we included 15 cases for the current review.6–20 Table 1 is a summary of their clinical features and outcomes.
| Clinical features | Number (%) |
|---|---|
| Total number of patients | 15 |
| Constitutional symptoms | 12 (80%) |
| Arthralgia | 03 (20%) |
| Pulmonary manifestation | 10 (66.67%) |
| Dyspnea | 10 (66.67%) |
| Asthma | 02 (13.33%) |
| Hemoptysis | 02 (13.33%) |
| Skin manifestation | 05 (33.33%) |
| Renal | 15 (100%) |
| Hematuria (microscopic) | 15 (100%) |
| Oliguria | 10 (66.67%) |
| Pyuria (microscopic) | 06 (40%) |
| Hematuria (Gross) | 01 (6.67%) |
| GI manifestation | 05 (33.33%) |
| Melena | 04 (26.67%) |
| Abdominal pain | 03 (20%) |
| Diarrhea | 01 (6.67%) |
| Neurological manifestation | 09 (60%) |
| Ophthalmological manifestation | 01 (6.67%) |
| ENT manifestation | 04 (26.67%) |
| Investigations | |
| Hemoglobin (mean±SD) gm/dl | 7.64 (± 2.72) |
| Platelet count, Median (IQR) per µl | 32,000 (18,000–50,000) |
| Fragmented RBCs, n (%) | 15 (100%) |
| High LDH, n (%) | 15 (100%) |
| Low haptoglobin1, n (%) | 14 out of 14 (100%) |
| Serum Creatinine, median(IQR) mg/dl | 5.2 (2.7–7.62) |
| Proteinuria, n (%) | 15 (100%) |
| Low C3 levels2, n (%) | 05 out of 11 (45.45%) |
| PR3 ANCA, n (%) | 05 (33.33%) |
| MPO ANCA, n (%) | 10 (66.67%) |
| Low ADAMTS3 (%) | 08 out of 10 (80%) |
| Abnormal genetic testing4 | 02 out of 03 (66.67%) |
| Abnormal chest imaging | 12 (80%) |
| Infiltrates | 09 out of 12 (75%) |
| Nodules | 03 out of 12 (25%) |
| Pleural effusion | 04 out of 12 (33.33%) |
| Treatment | |
| Pulse steroids | 14 (93.33%) |
| Oral steroids | 15 (100%) |
| IV cyclophosphamide | 04 (26.67%) |
| Oral cyclophosphamide | 03 (20%) |
| Rituximab | 04 (26.67%) |
| Azathioprine | 01 (6.67%) |
| Plasma exchange | 15 (100%) |
| Outcome | |
| Hematological improvement | 13 (86.67%) |
| Renal improvement | 08 (60%) |
| Mortality | 03 (20%) |
At presentation, the median age of the patients was 61 years (IQR 57–71 years), and 66.7% of them were females. Patients most commonly (80%) presented with constitutional symptoms like fever, malaise, and loss of appetite. Additionally, 10 patients (66.7%) experienced respiratory failure during their hospitalization [Table 2]. In eight patients, there was a simultaneous occurrence of TMA and AAV, whereas in others the latency between the two diagnoses ranged from one week to six months, with AAV preceding TMA. Most of the patients (66.7%) were MPO-ANCA-positive. All the patients had laboratory parameters consistent with systemic TMA, including progressively worsening anemia and thrombocytopenia with elevated lactate dehydrogenase, low haptoglobin, and presence of schistocytes. None of the cases had a history of accelerated hypertension preceding development of TMA.
| Author and year | Age | Sex | Country | Constitutional symptoms | Arthralgia | Dyspnea | Asthma | Hemoptysis | Rashes |
|---|---|---|---|---|---|---|---|---|---|
| Manabe et al., 20176 | 59 | M | Japan | Yes | No | Yes | No | Yes | No |
| Sathe et al., 20167 | 12 | M | India | Yes | Yes | Yes | No | No | Yes |
| Duong et al., 20208 | 82 | F | USA | Yes | Yes | Yes | No | No | No |
| Badiola et al., 20189 | 57 | M | Spain | Yes | Yes | Yes | Yes | No | Yes |
| Fukui et al., 201510 | 71 | M | Japan | No | No | Yes | Yes | No | Yes |
| Watanbe et al., 201011 | 61 | F | Japan | No | No | Yes | No | No | No |
| Agrawal et al., 201112 | 17 | F | USA | Yes | No | No | No | No | No |
| Asamiya et al., 200213 | 66 | F | Japan | Yes | No | Yes | No | No | No |
| Stefanidis et al., 199814 | 68 | F | Germany | Yes | No | Yes | No | No | No |
| Nagai et al., 200815 | 77 | F | Japan | Yes | No | No | No | No | No |
| Yamazaki et al., 200716 | 61 | F | Japan | Yes | No | No | No | No | Yes |
| Yamauchi et al., 201017 | 59 | F | Japan | Yes | No | No | No | No | Yes |
| Lim et al., 199818 | 66 | F | South orea | No | No | Yes | No | No | No |
| Shukla et al., 202219 | 36 | M | India | Yes | No | Yes | No | Yes | No |
| Kitamura et al., 202220 | 84 | F | Japan | Yes | No | No | No | No | No |
| Author and year | Oliguria | Hematuria | Abdominal pain | Melena | Diarrhea | Neurological manifestation | Neurological symptoms | Eye involvement | ENT involvement |
| Manabe et al., 20176 | Yes | No | No | No | No | No | No | No | No |
| Sathe et al., 20167 | No | Yes | No | No | No | No | No | No | No |
| Duong et al., 20208 | Yes | No | No | No | No | No | No | No | No |
| Badiola et al., 20189 | No | No | No | No | No | Yes | Paresthesia | No | Sinusitis |
| Fukui et al., 201510 | No | No | No | No | No | No | No | No | Sinusitis |
| Watanbe et al., 201011 | Yes | No | Yes | Yes | No | Yes | Speech and movement disturbances | No | No |
| Agrawal et al., 201112 | No | No | No | No | Yes | No | No | No | No |
| Asamiya et al., 200213 | Yes | No | No | Yes | No | Yes | Headache, emotional disturbances, and personality disorder | No | No |
| Stefanidis et al., 199814 | Yes | No | No | No | No | Yes | Headache | No | No |
| Nagai et al., 200815 | Yes | No | No | No | No | Yes | Paralysis and sensory disturbances of left side | No | No |
| Yamazaki et al., 200716 | Yes | No | Yes | Yes | No | No | No | No | No |
| Yamauchi et al., 201017 | Yes | No | No | Yes | No | Yes | Multiple mononeuritis and psychosis | No | No |
| Lim et al., 199818 | Yes | No | Yes | No | No | Yes | Deafness and facial palsy | Yes | Nasal ulceration and deafness |
| Shukla et al., 202219 | Yes | No | No | No | No | Yes | Left parieto-occipital bleed | No | Nasal ulceration |
| Kitamura et al., 202220 | No | No | No | No | No | Yes | Glove-and-stocking sensory loss | No | No |
M=Male; F=Female; ENT=Ear, nose, and throat
Five out of 11 patients (45.45%) had hypocomplementemia. Among the 10 patients tested for ADAMTS13 levels, 8 (80%) had low ADAMTS13 levels. However, only one patient (10%) had levels below 10% (Patient No. 1), which is the cut-off value for confirming TTP diagnosis [Supplementary Table 1].21 The remaining reports mandate a better characterization and, in all probability, a further investigation for complement-mediated HUS. In addition to this, three patients (20%), including our case, underwent genetic testing (Patient No. 3, 14, and 15); two cases had pathogenic mutations that predisposed them to develop HUS, and no pathological mutation was identified in the third case [Supplementary Table 1]. Supplementary Table 1 describes laboratory investigations.
Kidney biopsy
A kidney biopsy was carried out in 11 cases. Histological changes in the biopsy varied depending on the timing of the kidney biopsy after the onset of the disease. The biopsy details are shown in Supplementary Tables 2 and 3. The hallmark lesions of ANCA-associated glomerulonephritis, such as crescents and glomerular tuft necrosis, were seen in 10 (90.9%) and 4 (36.36%) patients, respectively. One patient (9.09%) had inflammatory cells with debris in the arteriolar wall (Patient No. 1) [Supplementary Table 3]. One patient had demonstrable glomerular TMA (Patient No. 3) [Supplementary Table 3]. Two patients (18.18%) had features suggestive of microangiopathies (vascular), such as endothelial swelling, fragmented red blood cells and leukocytes in the arteriolar wall, and myxoid changes in the intimal layer. One patient (9.09%) showed chronic changes of microangiopathy in the arterioles in the form of intimal thickening with near-complete occlusion of its lumen. In five patients, there were no changes of microangiopathy in the biopsy. However, all patients underwent a kidney biopsy before the onset of systemic TMA; the basis for diagnosing coexistent TMA and AAV was solely on clinical and other laboratory parameters.
Treatment and outcome
All patients underwent plasma exchange (PLEX). Seven (46.67%) received cyclophosphamide (either intravenously or oral). Three patients received rituximab as an induction agent (Patient No. 2, 3, and 15) [Supplementary Table 4].7,8,20 Additionally, one patient received rituximab and one patient received eculizumab for resistant disease: both responded favorably.13,20 Hematological remission occurred in 13 patients (86.67%), whereas 9 patients (60%) had kidney recovery with dialysis independence. In-hospital mortality occurred in three patients (20%). Two patients died of gut perforation, out of which one was attributed to gastrointestinal CMV infection. Our patient died due to an intracranial hemorrhagic event.19
Discussion
This case-based review highlights clinical features and renal outcomes of patients with systemic TMA (HUS or TTP) and AAV. The AAV preceding the TMA suggests the former as a probable trigger for the onset of TMA. Admittedly, the kidney outcome was dismal in patients with a synchronous presentation of AAV and TMA.
AAV is a multisystem disease. Constitutional symptoms, like fever and malaise, are common and precede organ-specific symptoms. To begin with, most patients (80%) in the current review had similar complaints. Upper respiratory tract involvement was frequent in PR3-AAV. Consistent with previous studies, lung involvement was present in 80% of patients: more in PR3-positive (100%) than in MPO-positive (70%) patients.22,23 Similar to a study by Mohammad et al.22 on 129 AAV patients, the most frequent lung parenchymal finding in this review was nodules in PR3-AAV and interstitial pneumonia in MPO-AAV patients. Cerebrovascular events are rare, and infarctions are more common than hemorrhage.24 Our previously published case report was one of few reports that demonstrated intracranial hemorrhage in AAV.19
Extra-renal manifestations are seen in 20% of patients with TMA and can involve the central nervous system (CNS) and cardiovascular and gastrointestinal systems. Renal manifestations in TMA are variable; however, most HUS patients are dialysis-dependent at presentation. TTP, in contrast to atypical HUS, uncommonly causes severe acute kidney injury.21
Until recently, the pathogenesis of AAV and HUS were considered discrete. However, there are a few explanations that may elucidate the rare co-occurrence of both diseases in the same individual. Firstly, pathogenesis of both the conditions involves the same target cell, that is, the endothelial cell. Secondly, emerging evidence from clinical and animal experiments suggests that the complement system is critical for developing AAV.25–27 C5a, an anaphylatoxin and chemoattractant, is pivotal for the pathophysiology of tissue injury in AAV, and therapeutic blockade of C5a effectively reduces the dose of corticosteroids in AAV with kidney involvement.28 One-third of the patients with AAV have hypocomplementemia, which is associated with poor kidney outcomes. Therefore, abnormal systemic activation of the alternate complement system is involved in the pathogenesis of both diseases. Third, the production of ANCA antibodies in AAV is associated with the formation of neutrophil extracellular traps (NETs).29 These NETs can act as a second hit in precipitating acute attacks of TMA in patients at risk of TMA.30 However, we all agree that in AAV, the evidence for dysregulation of the complement cascade is lacking and is limited to only tissue injury. However, beyond the complement system, there is yet to be a comprehensive explanation for both clinical conditions. Finally, like HUS, TTP may also coexist with AAV, though rarely. The endothelial damage in AAV causes the release of ultralarge von Willebrand factor (ULvWF) multimers from the endothelium.12 The imbalance between ULvWF and ADAMTS13 activity occurs when massive endothelial injury releases excessive ULvWF multimers, and this surpasses the cleaving capacity of ADAMTS13 and precipitates thrombosis.31 In spite of these potential multifactorial explanations, the exact mechanism of this co-occurrence is unclear and requires further evaluation.
In the cases reviewed here, TMA was either simultaneous or succeeding the presentations of AAV. Also, the presence of crescents in a significant number of glomeruli and lack of microangiopathic changes in the renal biopsy indicate AAV as the dominant pathology in such coexistence. Though vasculitic changes are the hallmark findings in AAV, thrombi and microangiopathic changes rarely occur in the arterioles. Microangiopathic changes in the arterioles (of the kidney) might intimate further evaluation for ADAMTS13 levels and complement regulatory proteins/autoantibodies (to CFH or ADAMTS13) or genetic testing in a patient with AAV.
Current guidelines (2021) from the American College of Rheumatology recommend patients with active, severe disease to be treated with rituximab over cyclophosphamide for remission induction to avoid adverse effects like neutropenia, bladder injury, and risk of infertility, even though both provide similar benefits in control of disease activity.32 Glucocorticoids are used simultaneously, beginning with high doses, typically intravenous methylprednisolone (500–1000 mg for three days) and followed by oral prednisolone. Following the PEXIVAS trial, the role of PLEX for severe AAV in preventing death or ESKD is debatable.33 On the contrary, PLEX remains the important treatment option for TMA.34 The presence of anti-ADAMTS13 antibodies and anti-CFH antibodies warrant immunosuppression. Current evidence strongly recommends the early use of eculizumab; however, we often encounter difficulty in a prompt and definitive diagnosis of atypical HUS and laboratory differentiation from TTP.35 Currently, these two diseases have different management approaches; diagnosing them appropriately while they coexist is crucial.
In terms of adverse renal outcomes of AAV, 59%–80% of AAV with severe renal involvement at presentation are dialysis-independent at 12 months.36 The mortality rate in the current review is consistent with prior studies of AAV and TMA. However, compared to previous studies of TMA and AAV, the incidence of ESKD was numerically higher (40%).5
Our review had certain limitations: small sample size, unavailability of kidney biopsy in all patients, and incomplete or inadequate workup for HUS and TTP, to name a few.
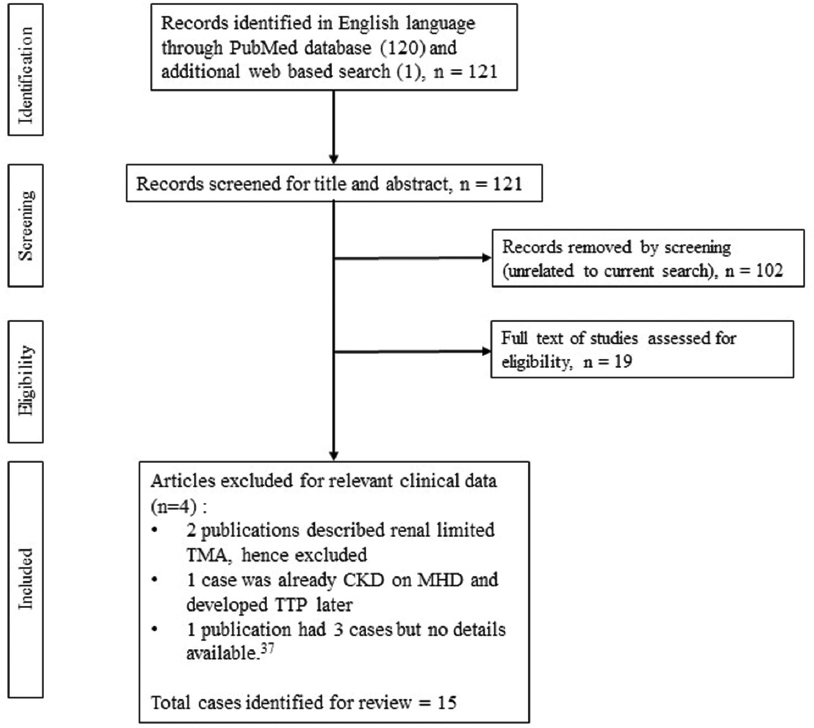
- Search strategy algorithm37. CKD: chronic kidney disease, MHD=maintenance hemodialysis, TMA=thrombotic microangiopathy.
Conclusion
Coexistence of AAV and TMA are uncommon, and the prognosis is abysmal. Given the rarity of this coexistence, we recommend a comprehensive workup and examination to provide additional insights into the disease.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
Nil.
References
- Classification of antineutrophil cytoplasmic autoantibody vasculitides: The role of antineutrophil cytoplasmic autoantibody specificity for myeloperoxidase or proteinase 3 in disease recognition and prognosis. Arthritis Rheum. 2012;64:3452-62.
- [CrossRef] [PubMed] [Google Scholar]
- Platelet counts as a biomarker in ANCA-associated vasculitis. Scand J Rheumatol. 2015;44:302-8.
- [CrossRef] [PubMed] [Google Scholar]
- Thrombotic microangiopathy in systemic lupus erythematosus: A cohort study in North Taiwan. Rheumatology. 2011;50:768-75.
- [CrossRef] [PubMed] [Google Scholar]
- Thrombotic microangiopathy associated with cryoglobulinemic membranoproliferative glomerulonephritis and hepatitis C. Am J Kidney Dis. 1998;31:521-6.
- [CrossRef] [PubMed] [Google Scholar]
- Clinicopathologic characteristics and outcomes of renal thrombotic microangiopathy in anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis. Clin J Am Soc Nephrol. 2015;10:750-8.
- [CrossRef] [PubMed] [Google Scholar]
- A case of PR3-ANCA-positive anti-GBM disease associated with intrarenal arteritis and thrombotic microangiopathy. CEN Case Rep. 2016;6:39-45.
- [CrossRef] [PubMed] [Google Scholar]
- Coexistence of atypical hemolytic uremic syndrome with membranoproliferative glomerulonephritis and antineutrophil cytoplasmic antibodies-associated vasculitis. Saudi J Kidney Dis Transpl. 2016;27:800-4.
- [CrossRef] [PubMed] [Google Scholar]
- Thrombotic microangiopathy and venous thrombosis in a patient with anti-neutrophil cytoplasmic antibody-associated vasculitis. Cureus. 2020;12:e11665.
- [CrossRef] [Google Scholar]
- Thrombotic microangiopathy in a patient with eosinophilic granulomatosis with polyangiitis: Case-based review. Rheumatol Int. 2019;39:359-65.
- [CrossRef] [PubMed] [Google Scholar]
- Eosinophilic granulomatosis with polyangiitis with thrombotic microangiopathy: Is simultaneous systemic lupus erythematosus associated with clinical manifestations? A case report and review of the literature. Medicine. 2015;94:e1943.
- [CrossRef] [PubMed] [Google Scholar]
- Thrombotic thrombocytopenic purpura in a patient with rapidly progressive glomerulonephritis with both anti-glomerular basement membrane antibodies and myeloperoxidase anti-neutrophil cytoplasmic antibodies. Clin Exp Nephrol. 2010;14:598-601.
- [CrossRef] [PubMed] [Google Scholar]
- Concomitant thrombotic thrombocytopenic purpura and ANCA-associated vasculitis in an adolescent. Pediatr Nephrol. 2011;26:1317-20.
- [CrossRef] [PubMed] [Google Scholar]
- Successful treatment with rituximab in a patient with TTP secondary to severe ANCA-associated vasculitis. Intern Med. 2010;49:1587-91.
- [CrossRef] [PubMed] [Google Scholar]
- Coincidence of haemolytic uraemic syndrome and c-ANCA-associated rapidly progressive glomerulonephritis. Nephrol Dial Transplant. 1998;13:1818-21.
- [CrossRef] [PubMed] [Google Scholar]
- Successful treatment of thrombotic thrombocytopenic purpura with repeated plasma exchange in a patient with microscopic polyangitis. Mod Rheumatol. 2008;18:643-6.
- [CrossRef] [PubMed] [Google Scholar]
- Wegener's granulomatosis complicated by intestinal ulcer due to cytomegalovirus infection and by thrombotic thrombocytopenic purpura. Intern Med. 2007;46:1435-40.
- [CrossRef] [PubMed] [Google Scholar]
- Successful treatment for thrombotic thrombocytopenic purpura complicated with myeloperoxidase anti-neutrophil cytoplasmic autoantibody-associated vasculitis. NDT Plus. 2010;3:279-81.
- [CrossRef] [PubMed] [Google Scholar]
- A case of Wegener's granulomatosis complicated by diffuse pulmonary hemorrhage and thrombotic thrombocytopenic Purpura. Korean J Intern Med. 1998;13:68-71.
- [CrossRef] [PubMed] [Google Scholar]
- Anti-neutrophil cytoplasmic antibody-associated vasculitis with haemolytic uraemic syndrome. Intern Med J. 2022;52:1450-1.
- [CrossRef] [PubMed] [Google Scholar]
- Anti-neutrophil cytoplasmic antibody-associated vasculitis complicated by thrombotic microangiopathy with posterior reversible encephalopathy syndrome successfully treated with eculizumab: A case report. Mod Rheumatol Case Rep. 2022;6:254-9.
- [CrossRef] [PubMed] [Google Scholar]
- How I treat: The clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome. Blood. 2014;123:2478-84.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary involvement in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: The influence of ANCA subtype. J Rheumatol. 2017;44:1458-67.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary manifestations of wegener granulomatosis: CT findings in 57 patients and a review of the literature. Eur J Radiol. 2005;53:471-7.
- [CrossRef] [PubMed] [Google Scholar]
- Central nervous system involvement in ANCA-associated vasculitis: What neurologists need to know. Front Neurol. 2019;9:1166. doi: 10.3389/fneur.2018.01166
- [CrossRef] [PubMed] [Google Scholar]
- Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2013;83:129-37.
- [CrossRef] [PubMed] [Google Scholar]
- Glomerular C3d as a novel prognostic marker for renal vasculitis. Hum Pathol. 2016;56:31-9.
- [CrossRef] [PubMed] [Google Scholar]
- Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52-64.
- [CrossRef] [PubMed] [Google Scholar]
- Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. 2021;384:599-609.
- [CrossRef] [PubMed] [Google Scholar]
- Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol. 2014;25:990-7.
- [CrossRef] [PubMed] [Google Scholar]
- Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120:1157-64.
- [CrossRef] [PubMed] [Google Scholar]
- Two cases of refractory thrombotic thrombocytopenic purpura associated with collagen vascular disease were significantly improved by rituximab treatment. Clin Rheumatol. 2007;26:2159-62.
- [CrossRef] [PubMed] [Google Scholar]
- 2021 American College of Rheumatology/vasculitis foundation guideline for the management of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Care Res. 2021;73:1088-105.
- [CrossRef] [PubMed] [Google Scholar]
- Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med. 2020;382:622-31.
- [CrossRef] [PubMed] [Google Scholar]
- Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol. 2012;158:323-35.
- [CrossRef] [PubMed] [Google Scholar]
- Atypical hemolytic uremic syndrome. Hematology Am Soc Hematol Educ Program. 2016;2016:217-25.
- [CrossRef] [PubMed] [Google Scholar]
- Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180-8.
- [CrossRef] [PubMed] [Google Scholar]
- Thrombotic microangiopathy associated with anti-neutrophil cytoplasmic antibody-associated vasculitis: A French nationwide retrospective case-control study and literature review. Rheumatology. 2019;58:1873-5.
- [CrossRef] [PubMed] [Google Scholar]




