

Translate this page into:
Atypical HUS Associated with CFH/CFHR-1 Hybrid Gene
Address for correspondence: Dr. Valentine Lobo, Renal Unit, Diamond Jubilee Building, KEM Hospital, Sardar Moodliar Road, Rasta Peth, Pune - 411 011, Maharashtra, India. E-mail: valentinelobo@rediffmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Atypical hemolytic uremic syndrome is a rare form of thrombotic microangiopathy caused by complement pathogenic variants. We describe a case of a 33-year-old woman who presented as rapidly progressing renal failure requiring dialysis and had anemia, microhematuria, low C3, normal C4 levels, and normal platelet count. Renal biopsy revealed arteriolar thrombotic microangiopathy and acute tubular injury. Patient was treated with plasma exchange and hemodialysis as required. This resulted in partial recovery at 1 month. Genetic workup by multiplex ligation-dependent probe amplification revealed a 1.5 times higher signal intensity on downstream region of CFH gene and 50% reduced intensity of exon 6 of CFHR1 gene, suggesting a gene conversion event, similar to those previously reported from Spain and Portugal.
Keywords
Atypical hemolytic uremic syndrome
multiplex ligand-dependent probe amplification assay
thrombotic microangiopathy

Introduction
Atypical hemolytic uremic syndrome (aHUS) is a rare disease characterized by acute kidney injury (AKI), thrombocytopenia, and microangiopathic hemolytic anemia (MAHA) with an incidence of approximately 0.5 per million per year.[1] Inherited and/or acquired complement abnormalities are reported in 50%–70% of patients with aHUS.[12] We report here a case of thrombotic microangiopathy (TMA) with an unusual variation in complement genes.
Case Presentation
A 33-year-old married woman with two previous uneventful pregnancies was admitted from the emergency department of our hospital with bilateral lower limb edema for 7 days, breathlessness at rest, and oliguria since 4 days. She had no complaints of hematuria, hemoptysis, malar rash, arthralgia, oral ulcers, photosensitivity, or hair loss. Her serum creatinine done 3 years earlier was 1.15 mg/dl. Prior to admission at our hospital, she was detected to have a serum creatinine of 17 mg/dl and had received two sessions of hemodialysis and 500 mg of intravenous methylprednisolone. At the time of admission, she was afebrile and had tachycardia. Her blood pressure of 140/90 mmHg and oxygen saturation was 94% on room air. Physical examination revealed jugular venous distension and bilateral lower limb edema. Examinations of oral cavity and respiratory system were within normal limits; there were no visible rashes. Investigations showed hemoglobin of 7.2 g/dl, platelet count of 240,000/μl with no schistocytes. Urine examination showed 3+ proteinuria, 8–10 red cells, and 50–55 leucocytes. Serum lactate dehydrogenase was 585 IU/L; serum levels of complement C3 were marginally low (88 mg/dl, Normal = 90–180 mg/dl) while C4 levels were normal (22 mg/dl, Normal = 10–40 mg/dl). Ultrasonogram showed bilateral swollen kidneys with preserved corticomedullary differentiation and normal parenchymal echogenicity. She was dialyzed on two consecutive days for pulmonary edema. Renal biopsy showed arteriolar TMA in acute phase with acute tubular injury. Immunofluorescence staining for IgG, IgM, IgA, C3, C1q, κ and λ was negative.
Serum anti-factor H antibody level was 495 AU/L (Normal range: 0–100 AU/L) and plasma ADAMTS-13 activity was normal (60%). Plasma exchange (PEX) with 4.2 L of volume for volume replacement with fresh frozen plasma was started for TMA, secondary to dysregulation of complement alternative pathway. She received five daily PEX till platelets and LDH normalized, followed by nine PEX on alternate days, along with hemodialysis as required. Urine output gradually increased and after one month the patient became dialysis-independent. She was discharged with a serum creatinine of 3.3 mg/dl. Follow-up 45 days after the diagnosis showed serum creatinine of 2.4 mg/dl.
Multiplex ligation-dependent probe amplification (MLPA) for copy number variation (CNV) of genes involved in alternate complement pathway was carried out [SALSA MLPA P236-A3 probemix, MRC Holland] on DNA extracted from the peripheral blood. The MLPA results, when compared to a sample from a known healthy control run in parallel, showed an ~1.5X increase in relative signal intensity for one probe targeting the terminal region of the CFH gene (exon 23) and a ~50% reduction in signal for one probe targeting exon 6 of the CFHR1 gene [Figure 1]. These findings suggest a duplication and deletion in these regions, respectively, which in turn, indicate a gene conversion event between the CFH and CFHR1 genes. Clinical exome sequencing using the Illumina Trusight One platform did not detect any clinically relevant sequence variant in any of the complement pathway related genes (CFH, CFI, CD46/MCP, C3)or ADAMTS13. A functional assay for factor H as previously described by Sanchez Coral et al.[3] was performed. It showed a 33% lysis of sheep erythrocytes as compared to 5% with control plasma from healthy blood donors. The healthy donor plasma also inhibited the lysis inducedin vitro by the patient's plasma in a dose-dependent manner [Figure 2].

- MLPA report. The ratio of the signal intensity of the test sample to the normal control in the capillary electrophoretogram on multiplex ligation-dependent probe amplification (MLPA) testing. The terminal exon of FH gene shows a 1.5 times increased signal intensity and exon 6 of CFHR1 gene shows a 50% reduction. This suggests 3 copies of exon 23 of factor H and 1 copy of exon 6 of CFHR1 gene are detected, possibly due to a gene conversion event
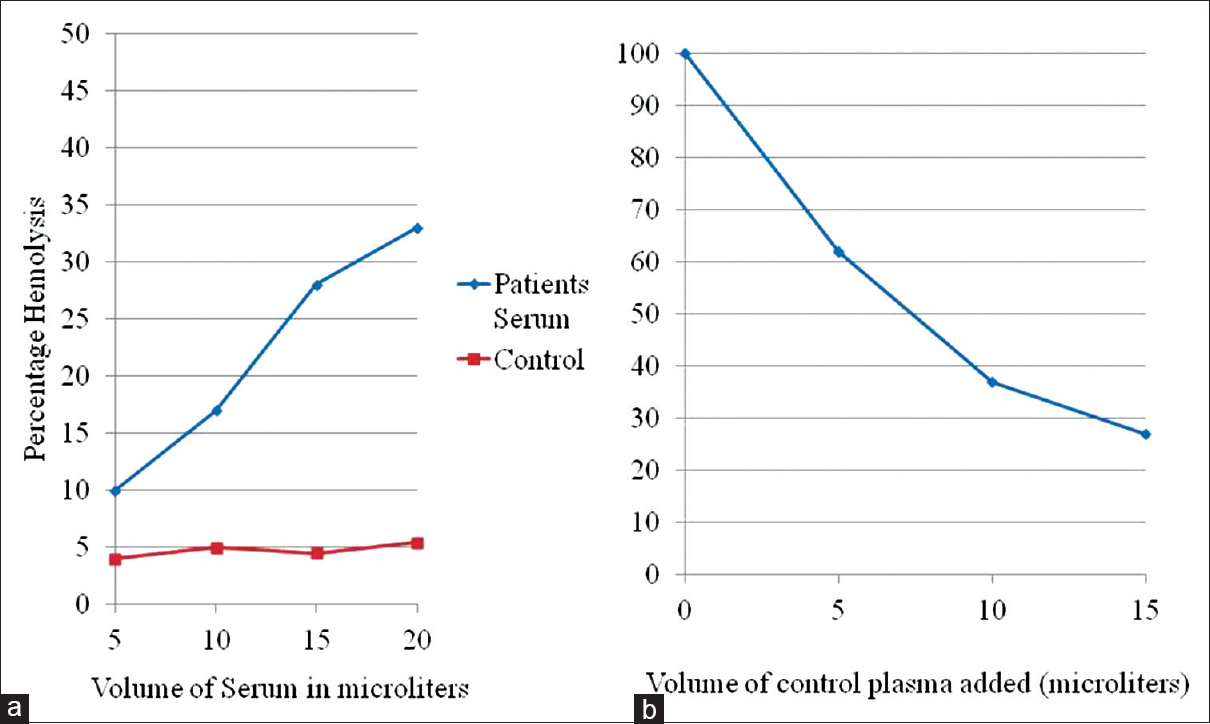
- Functional assay for factor H by in vitro sheep erythrocyte lysis. (a) On adding from 5 to 20 μl of the patient's plasma to EGTA buffer, there was a progressive increase in percentage lysis up to 33% compared with 5% for healthy donor plasma used as control. (b) When patient's plasma was mixed in serial dilutions with control plasma, there was a dose-dependent decrease in lysis indicating a competition by control factor H with the hybrid protein
Discussion
TMA defines a spectrum of disorders characterized by thrombocytopenia and MAHA due to endothelial injury.[4] The pathophysiology of aHUS is assumed to be due to the excessive activation of the alternative complement pathway. Germline variants in genes involved in alternate complement pathway may be associated with an unchecked activation of alternative complement pathway.[5] These variants include loss-of-function mutations in complement factor H (CFH), complement factor I (CFI), membrane cofactor protein, (MCP/CD46), gain-of-function mutations in C3 and complement factor B (CFB) genes, CNV's in complement factor related genes (CFHR 1–5), or a combination of the above.
Fluid phase and surface anchored complement proteins combine to limit complement activation with factor H being the most important fluid-phase regulator. The most frequently reported genomic variants associated with aHUS are heterozygous variants in CFH gene, which are observed in 21%–25% of cases.[6] The most of the clinically relevant variants in CFH gene are found in the C-terminal region coded by exon 23 and involving short consensus repeats (SCRs) 19 and 20, which is responsible for endothelial cell binding and surface activity while the N-terminal end comprising SCR 1–7 contains enzyme cofactor activity. Thus, mutations in SCRs 19 and 20 result in a loss of surface binding and unregulated complement activity on endothelial surfaces with formation of the membrane attack complex, microvascular thrombosis, and organ damage.[7] CFHR proteins 1 to 5 are structurally similar to factor H, the coding regions of which are located next to the CFH gene on chromosome 1. CFHR genes, considered to be pseudogenes, carry a high degree of sequence homology with CFH and hence are susceptible for genetic rearrangements leading to the formation of CFH-CFHR hybrids.[8] The two Cterminal SCR domains of CFHR1–5 are 95% homologous to the factor H Cterminus. However, CFHR1 lacks the regulatory function of factor H which requires N-terminus.[8] All variants reported in CFH and CFHR1–5 show around 50% penetrance both in familial and sporadic cases and require a second hit or trigger for development of the disease in susceptible individuals carrying the mutation.[9]
CFH –CFHR hybrid genes are rare, contributing to 1%–5% of all aHUS cases.[8910] An Italian study reported the incidence of such hybrids in 1% of their 273 aHUS patients.[7] Bresin et al. reported one case with CFH-CFHR hybrid among 795 aHUS patients.[11] Similarly, Eyler et al. in 2013 reported aHUS due to a CFHR1/CFH fusion protein.[12] A recent study by de Jorge et al. identifled 9 patients carrying a novel genetic abnormality that resulted in the fusion of SCRs 1–3 of CHFR1 with SCRs 19–20 of CFH in their aHUS cohort of 513 patients.[2] The authors noted that this genetic abnormality escaped detection by next-generation massive parallel DNA (NGS) sequencing, but was detected as CNV by MLPA. In addition, Sanger sequencing in 6 of these patients revealed two genetic variants in heterozygosis causing the amino acid substitutions, L290S and A296V in CFHR1 exon 6 and heterozygosity for the S411T genetic variant in exon 9 of CFH. Sanger sequencing in one patient showed a heterozygosity for CFHR1 exon 6 causing the amino acid substitutions, L290S and A296V, while even Sanger sequencing did not reveal any abnormality in the remaining 2 patients. MLPA in our patient suggested a gene conversion event involving exon 23 of CFH gene and exon 6 of CFHR1 gene and we could not perform Sanger sequencing for our patient.
The hybrid gene identified in our patient is identical to the CFHR1: CFH hybrid gene described by de Jorge et al.,[2] Valoti et al.,[13] and Eyler et al.,[12] in which the contribution of this variant to the overall incidence of HUS was 4.3%. These authors have reported a disease penetrance of 43%, poor response to therapies other than Eculizumab, poor prognosis, and high risk of recurrence after transplantation. They also reported that the disease was influenced markedly by the ratio of the normal FH to that of the mutant protein, other genetic variants, and environmental triggers, none of which could be identified in our patient probably because of the late presentation to us.
The protein formed by transcription of the mutant CFHR1 gene would have a binding site with 100% homology with that of CFH but would lack regulatory activity. The presence of a competitor to normal factor H activity is also suggested by thein vitro lysis of sheep erythrocytes by the patient's plasma which was inhibited competitively by factor H from normal control plasma.
While 57% of the affected women in the Spanish cohort[2] first came to attention with post-partum HUS, our patient had been through two uneventful pregnancies and we could not identify a triggering event for her disease.
The limitation of the case report is the lack of confirmation of the presence of a hybrid gene which could have been achieved by a custom-designed assay targeting the CFH and CFHR1 genes. Because of the high degree of homology between CFH and CFHR1 genes and short-read sequencing of only coding region, the presence of hybrid gene could not be detected by capture-based commercial NGS Illumina TruSight One panel. Sanger sequencing which yields a higher frequency of mutation could not be performed and the phenotype-genotype correlation and penetrance could not be completely established as no family members were willing to undergo screening.
In conclusion, TMA without systemic manifestations is a challenging condition, requiring a high index of clinical suspicion and renal biopsy for definitive diagnosis. The genetic abnormality we reported is a rare phenomenon, which could only be suggested by MLPA. The presence of hybrid gene can be confirmed by custom targeted sequencing or long-read sequencing (Sanger sequencing) and not by capture-based short-read sequencing used by most of the commercial NGS panels. The renal biopsy findings, in the absence of usual clinical and laboratory features of a TMA, led to early initiation of PEX and partial renal recovery in our patient.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understands that her name and initials will not be published and due efforts will be made to conceal her identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Conference Participants. Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int. 2017;91:539-51.
- [Google Scholar]
- Factor H competitor generated by gene conversion events associates with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2018;29:240-9.
- [Google Scholar]
- Functional analysis in serum from atypical Hemolytic Uremic Syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol. 2004;41:81-4.
- [Google Scholar]
- Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21:895-67.
- [Google Scholar]
- Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844-59.
- [Google Scholar]
- Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: Core curriculum 2015. Am J Kidney Dis. 2015;66:359-75.
- [Google Scholar]
- Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24:475-86.
- [Google Scholar]
- A novel hybrid CFHR1/CFH gene causes atypical hemolytic uremic syndrome. Pediatr Nephrol. 2013;28:2221-5.
- [Google Scholar]
- A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol. 2015;26:209-19.
- [Google Scholar]




