

Translate this page into:
Bones, Stones, and Hematuria - Connecting the Dots
Corresponding author: Madhura P. Fadnis, Bramha 503, Om Trimurti Society, Sinhagad Road Pune, Maharashtra, India. E-mail: madhura.fadnis1@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Fadnis MP, Kalrao V, Kalra S, Acharya S. Bones, Stones, and Hematuria - Connecting the Dots. Indian J Nephrol. 2025;35:294-6. doi: 10.25259/ijn_531_23
Abstract
We report a 12 yr old boy who presented with recurrent gross hematuria, polyuria and rickets with growth failure. Investigations showed bilateral renal calculi with small kidneys on ultrasonography along with hypercalciuria; hypomagnesemia and reduced kidney function. His younger sibling also had nephrocalcinosis hypomagnesemia. The genetic analysis done in view of recurrent renal calculi with chronic kidney disease showed a homozygous missense variant (c.392G>A) at exon 4 of CLDN 16 gene suggestive of Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC). The younger sibling had a similar homozygous mutation and the parents were heterozygous carriers.
Keywords
FHHNC
Hypomagnesemia
Hypercalciuria
Recurrent gross hematuria
Rickets

Introduction
There are various causes of recurrent kidney stones in pediatric population, such as adenine phosphoribosyltransferase (APRT) deficiency, cystinuria, Dent disease, familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC), and primary hyperoxaluria (PH).1 Lack of recognition can result in delays in treatment. Here, we report a 12-year-old boy with recurrent gross hematuria, polyuria, and rickets with growth failure, with a similar history in a younger sibling.
Case Report
A 12-year-old boy, the first born of a nonconsanguineous marriage, presented with a history of recurrent gross painless hematuria since last 4 years. There was a history of nocturia and constipation in the child from early infancy with poor weight and height gain and bowing of the legs. Two years back, the child had sustained a fracture of the distal end of the radius on trivial trauma. His investigations had shown severe hypocalcemia with radiological features of rickets and renal calculi on ultrasound of the kidneys. The younger sibling also had a history of painless gross hematuria.
Examination revealed weight and height below the third centile as per the Indian Academy of Pediatrics (IAP) growth charts.2 Pallor and bilateral genu valgum were present. His investigations showed deranged renal function tests with hypocalcemia, hypomagnesemia, and hypercalciuria [Table 1]. There was bilateral nephrolithiasis on ultrasound of the kidneys and urinary tract [Figure 1]. The laboratory investigations in the younger sibling showed similar results, except that he had an estimated creatinine clearance of 75 ml/min/m2.
| Investigations | Index patient | Younger sibling |
|---|---|---|
| Hemogram | 9.8/6400/2.9 | 10 |
| Urine routine |
RBC 15–20 WBC 10–15 |
RBC WBC |
| Serum electrolytes | 137/3.8/116 | 140/4 |
|
Serum urea Serum creatinine |
49/2.23 eGFR: 24 |
20/0.6 eGFR: 75 |
| VBG | 7.21/34/13.6 | 7.26/36/14 |
| UCCR | 0.7 mg/mg | 0.9 |
| UPCR | 2.54 mg/mg | 1.8 |
| Serum Mg | 1.8 (normal range {NR}: 1.7–2.2) | 0.9 |
| Serum Ca/PO4/ALP | 7.6/3.6/307 | 8.7/3.8/290 |
| Vit. D3/serum parathyroid hormone PTH | 30/189 (NR: 70–110) | 28/190 |
ALP: Alkaline phosphatase, eGFR: Estimated glomerular filtration rate, RBC: Red blood cells, WBC: White blood cells, NR: Normal range, Ca: Calcium, PO4: Phosphorus, ALP: Alkaline phosphatase, PTH: Parathormone, UCCR: Urine calcium creatinine ratio, UPCR: Urine protein creatinine ratio, VBG: Venous blood gas, Mg: Magnesium.
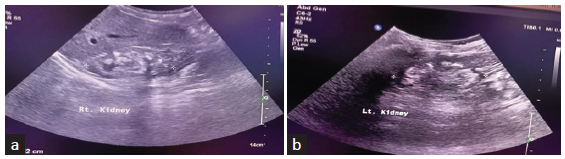
- (a and b) Ultrasonographic images of both the kidneys of the index patient showing small-sized kidneys with bilateral nephrolithiasis.
Suspecting an underlying genetic etiology, whole exome sequencing was ordered, which showed a novel homozygous missense mutant C.392G>A at exon 4 of CLDN 16 gene, suggestive of FHHNC. Sanger sequencing showed a heterozygous mutation in parents and a homozygous mutation at a similar location in the younger sibling [Figure 2].
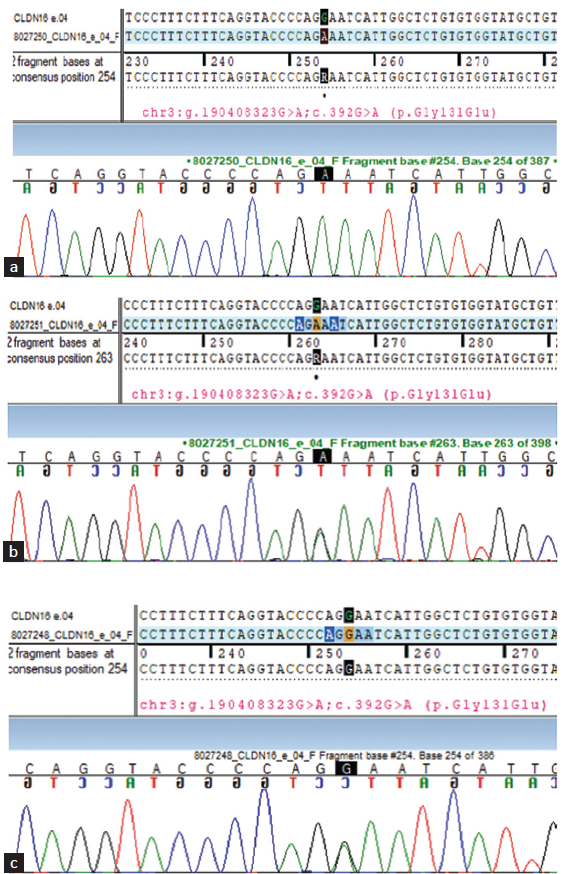
- (a) Sanger sequencing of the younger sibling. (b) Sanger sequencing of the mother. (c) Sanger sequencing of the father. Colored lines indicate different nucleotides in the DNA sequence: red - Nucleotide T, green - Nucleotide A, blue - Nucleotide C, black - Nucleotide G.
Discussion
Children with recurrent stone disease have been reported to have underlying metabolic cause with progression to chronic kidney disease (CKD).3 Our patient was detected to have FHHNC by whole exome sequencing.
FHHNC, a rare autosomal recessive disease is of two types I and II, which occur due to mutation of CLDN 16 and CLDN 19 genes present on chromosomes 3q27 and 1p34.2, respectively. The mutations are responsible for the defective production of claudin proteins 16 and 19, respectively.4 These proteins that are expressed in the tight junction of the thick ascending limb of the loop of Henle play an important role in the paracellular transport of calcium and magnesium with the help of electropositive luminal gradient facilitated by sodium potassium chloride cotransporter (NKCC2) and renal outer medullary potassium (ROMK) channels and Na/K ATPase pump.
The clinical presentation can be in the form of polyuria, nephrolithiasis, recurrent urinary tract infections, growth retardation, and rickets, similar to that observed in our index patient. Investigations show hypomagnesemia with magnesuria and hypercalciuria. Other findings include nephrocalcinosis, mild normal anion gap metabolic acidosis, hyperuricemia, and hyperparathyroidism, which are characteristically inappropriate to the degree of CKD and hypomagnesemia. About one-third of children can progress to end-stage renal disease (ESRD) by the second decade of life.4 Our patient had normal magnesium levels with progression to CKD IV. This can be because of the advanced stage of the kidney disease, which results in decreased filtered load of magnesium reaching the tubules. The younger sibling showed a low serum magnesium level of 0.9 mg/dl.
In our patient, a novel missense variant was identified at exon 4 of CLDN 16 gene. The mutation occurs due to the substitution of glutamic acid by glycine at codon 131 (p.Gly131Glu). The variant has not been reported in 1000 genomes, gnomAD (v3.1), gnomAD (v2.1), and TOPMed databases and has a minor allelic frequency of 0.0009% in our databases. The younger sibling also had a similar mutation at exon 4. This is the first case report from India with a novel mutation. Where a complete genetic evaluation has been done of both the siblings and the parents to confirm the diagnosis of FHHNC.
A previous report identified this mutation in exon 2.5 A study from AIIMS identified another novel mutation in two unrelated patients.6 Another case report showed a mutation c.620G>A at exon 4 in one of the siblings.1 Treatment involves magnesium and citrate supplementation and thiazides to reduce hypercalciuria. However, kidney transplant is the definitive treatment.
Our case report highlights the need to thoroughly investigate children with recurrent kidney stones as many can have underlying metabolic and genetic defects which can lead to progression to CKD.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
References
- Familial hypomagnesemia, hypercalciuria and nephrocalcinosis with novel mutation. Indian J Nephrol. 2019;29:57-61.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Pediatrician-friendly IAP growth charts for children aged 0-18 years. Indian Pediatr. 2020;57:997-8.
- [PubMed] [Google Scholar]
- Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013;28:1923-42.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis: Clinical and molecular characteristics. Clin Kidney J. 2015;8:656-64.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- POS-084 a rare genetic disorder presenting as recurrent urinary tract infection in children. Kidney Int Rep. 2022;7:s35.
- [Google Scholar]
- Novel pathogenic variant causing familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Asian J Pediatr Nephrol. 2020;3:31.
- [Google Scholar]




