

Translate this page into:
Co-existence of classic familial lecithin-cholesterol acyl transferase deficiency and fish eye disease in the same family
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
We report a family with a rare genetic disorder arising out of mutation in the gene that encodes for the enzyme lecithin-cholesterol acyltransferase (LCAT). The proband presented with nephrotic syndrome, hemolytic anemia, cloudy cornea, and dyslipidemia. Kidney biopsy showed certain characteristic features to suggest LCAT deficiency, and the enzyme activity in the serum was undetectable. Mother and younger sister showed corneal opacity and dyslipidemia but no renal or hematological involvement. These two members had a milder manifestation of the disease called fish eye disease. This case is presented to emphasize the importance of taking family history and doing a good clinical examination in patients with nephrotic syndrome and carefully analyze the lipid fractions in these subset of patients.
Keywords
Corneal opacity
dyslipidemia
lecithin-cholesterol acyltransferase
nephrotic syndrome

Introduction
Familial lecithin-cholesterol acyltransferase deficiency (FLD) and fish eye disease (FED) are autosomal recessive disorders arising out of mutation in the LCAT gene, which encodes for the enzyme lecithin-cholesterol acyltransferase. Patients with classic FLD develop nephrotic syndrome progressing to end-stage renal disease (ESRD), corneal opacities, dyslipidemia and hemolytic anemia. FED is a milder form of the disease associated with corneal opacity and dyslipidemia. We report a family where one member was affected with classic FLD while two others were found to have FED [Figure 1].
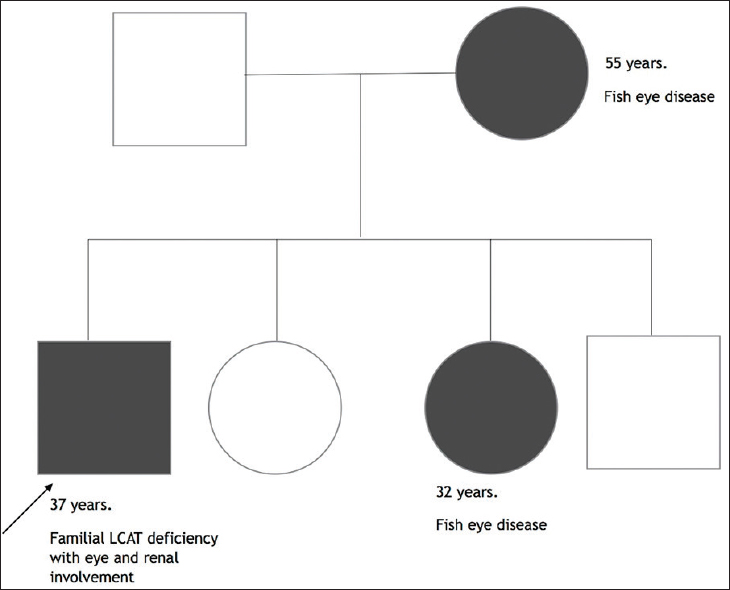
- Pedigree chart of the family depicting the affected members of the family with lecithin-cholesterol acyltransferase deficiency or fish eye disease
Case Report
A 37-year-old male presented with progressive swelling of feet and facial puffiness for 6 months. On physical examination, he had mild pallor, moderate pedal edema and cloudy cornea with a peripheral arcus in both eyes [Figure 2].

- Clinical and histological images of the patient with lecithin-cholesterol acyltransferase (LCAT) deficiency. Panel a - Photograph showing the cloudy cornea of the patient characteristic of LCAT deficiency. Panel b - Photomicrograph of H and E, stained kidney biopsy specimen (×40) demonstrating mesangial expansion with thickening of glomerular capillary loops and vacuolization of basement membrane (solid arrow). Panel c - Photomicrograph of silver stained kidney biopsy specimen (×40) demonstrating thickened glomerular basement membrane
Laboratory investigations revealed hemoglobin of 8 g/dL with “target” erythrocytes in peripheral blood film examination; serum haptoglobin of 64 mg/dl (normal range: 70–200 mg/dl), total cholesterol 215 mg/dl, high-density lipoprotein cholesterol (HDL-C) 10 mg/dl, and triglycerides 385 mg/dl. Renal biopsy showed glomerulomegaly with mild mesangial expansion and foam cell infiltration in mesangium and glomerular basement membrane (GBM) [Figure 2]. Tubules also showed foamy changes with focal tubular atrophy, and interstitium showed the presence of foam cells.
In view of the clinical profile, characteristic dyslipidemia (high triglyceride, very low HDL) and histological changes, serum LCAT deficiency were suspected. Plasma LCAT activity was determined at Pacific Biomarkers, Seattle, USA. The cholesterol esterification rate was undetectable indicating complete lack of LCAT in plasma. Clinical evaluation of family members revealed corneal opacities and dyslipidemia in patient's mother and his younger sister. However, there was no renal, hematological or other systemic involvement in both of them. Genetic testing of the proband and serum LCAT activity of the other affected members could not be done due to financial constraints. Based on the clinical manifestations, the proband was diagnosed to have classic FLD while his mother and sister were diagnosed to have FED.
Discussion
Familial LCAT deficiency is an autosomal recessive disorder caused by mutations in LCAT gene, which is located in the chromosome 16q22.[1] It has a prevalence of <1:1,000,000 and about 70 families with this condition have been reported worldwide.[2] The disorder was first reported in 1967 in a Norwegian family.[3] The mutation underlying this disorder was identified in 1992[4] The disorder is characterized clinically by corneal opacity, hemolytic anemia, proteinuria and renal dysfunction. ESRD is the major cause of morbidity and mortality in this condition. The first case of LCAT deficiency from India was reported by Muthusethupathi et al.[5] wherein two siblings were found to be affected with this disease, and one of them developed ESRD requiring renal transplant. The mean age of ESRD is usually in the fourth decade of life.
Lecithin-cholesterol acyltransferase is an enzyme that is expressed primarily in the liver and secreted into the plasma compartment.[6] The enzyme was first identified[7] in 1962. The enzyme plays an important role in esterification of free cholesterol in lipoproteins leading to maturation of HDL that helps in reverse cholesterol transport.[89] The esterification of free cholesterol by LCAT primarily occurs at the surface of HDL, and this is called alpha-LCAT activity.[10] However, about 25% of LCAT activity is also detected in other lipoproteins such as low-density lipoprotein and very low-density lipoprotein, called beta-LCAT activity.[8] Thus, alpha and beta LCAT activities are two functional aspects of the same protein.[10] There are two clinical syndromes that arise out of mutations in LCAT gene namely – FLD and FED.
In FED, there is a loss of alpha-LCAT activity while the beta-LCAT activity is preserved.[11] Individuals with FED have very low level of HDL and corneal opacities.
Classic FLD is characterized by complete lack of alpha and beta LCAT activity. Therefore, the unesterified cholesterol accumulates in all plasma lipoproteins. The patients may present with HDL-C deficiency, corneal opacification, hemolytic anemia, hypertension, hypertriglyceridemia, and proteinuria frequently progressing to ESRD.[1011] The difference in clinical and bio-chemical features between FLD and FED is summarized in Table 1. The renal involvement in FLD is mediated by deposition of lipoprotein X particles mainly in the glomeruli.[12]

Light microscopic examination of kidney biopsy usually reveals mild mesangium expansion with thickening of GBM, deposition of foamy lipids in the thickened GBM. Similar findings are also observed in the interstitium and blood vessel wall. Careful examination of the biopsy specimen is crucial, as cholesterol deposition may sometimes be focal and can be missed. Frascà et al. reported a case of LCAT deficiency wherein the findings were missed in a patient's initial two kidney biopsies and a third kidney biopsy picked up the foam cell deposition in various compartments of kidney tissue.[13] Corneal opacification in LCAT deficiency due to phospholipid accumulation usually starts early in life and often is the presenting symptom of this disease.[14] Anemia in these patients is caused by unesterified free cholesterol and phosphatidylcholine deposition in erythrocyte membrane.[12]
The disease is to be suspected whenever a young patient with nephrotic syndrome has the following constellation of clinical and biochemical parameters - cloudy cornea, disproportionate anemia (hemolytic), hypertension, dyslipidemia with high triglycerides and extremely low HDL and other findings as mentioned in Table 1. Clinical evaluation and urine examination of other family members are very important as significant phenotypic variations are seen within the same family carrying the same mutation. Classic biopsy findings support the diagnosis. However, definitive diagnosis requires measurement of LCAT activity that is generally undetectable in cases of classic LCAT deficiency and partially lost in patients with FED though exact cut-off levels are not available. Genetic testing helps in identifying the underlying mutations but is done only in highly specialised labs.
There is no definitive therapy for this disorder. Our patient is being conservatively managed with low fat and low salt diet, angiotensin converting enzyme inhibitor and statins. There is a theoretical role of gene therapy or liver transplantation in this disorder but so far they have not been tried. In patients who progress to ESRD, renal transplantation is the treatment of choice. Recurrence of the disease in the allografts is known to occur between few months to as late as 42 months posttransplant[1516] and progression to ESRD is apparently faster than in native kidneys.[17] But the recurrence rate is very unpredictable, and the disorder is not a contraindication for renal transplant. Whether combined liver-kidney transplant will prevent recurrence or not needs to be studied.
In the family that we have reported, the proband who was first evaluated satisfied the criteria for classic FLD while the patient's sister and mother satisfied the criteria for FED. It is interesting to note that despite being in the same family, (and probably having the same mutation), the clinical presentation and biochemical studies show significant phenotypic variability.
We report this case to highlight the importance of taking a detailed family history and doing a thorough physical examination in every case of the nephrotic syndrome. The disease should be suspected when a patient with the nephrotic syndrome has cloudy cornea, disproportionate anemia, very low HDL levels and a positive family history.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Genetics of LCAT (lecithin: Cholesterol acyltransferase) deficiency. Ann Hum Genet. 1975;38:327-31.
- [Google Scholar]
- Orphanet: Familial LCAT Deficiency. Available from: http://www.orpha.net/consor4.01/www/cgi.bin/OC_Exp.php?lng=EN and Expert=79293
- Familial plasma lecithin: Cholesterol acyltransferase deficiency biochemical study of a new inborn error of metabolism. Scand J Clin Lab Invest. 1967;20:231-43.
- [Google Scholar]
- The genetic defect of the original Norwegian lecithin: Cholesterol acyltransferase deficiency families. FEBS Lett. 1992;309:307-10.
- [Google Scholar]
- Familial lecithin: Cholesterol acyltransferase deficiency with renal failure in two siblings. First case report from India. Nephron. 1999;81:89-93.
- [Google Scholar]
- The molecular pathology of lecithin: Cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res. 1997;38:191-205.
- [Google Scholar]
- The mechanism of the plasma cholesterol esterification reaction: Plasma fatty acid transferase. Biochim Biophys Acta. 1962;65:128-35.
- [Google Scholar]
- Advances in understanding of the role of lecithin cholesterol acyltransferase (LCAT) in cholesterol transport. Clin Chim Acta. 1999;286:257-71.
- [Google Scholar]
- Familial LCAT deficiency and fish — Eye disease. 1988. Studies in Inherited Metabolic Disease. Netherlands: Springer; :45-56. Available from: http://link.springer.com/chapter/10.1007/978-94-009-1259-5_5
- [Google Scholar]
- Fish eye syndrome: A molecular defect in the lecithin-cholesterol acyltransferase (LCAT) gene associated with normal alpha-LCAT-specific activity. Implications for classification and prognosis. J Clin Invest. 1993;92:479-85.
- [Google Scholar]
- Histopathology of corneal changes in lecithin-cholesterol acyltransferase deficiency. Cornea. 2002;21:834-7.
- [Google Scholar]
- A 33-year-old man with nephrotic syndrome and lecithin-cholesterol acyltransferase (LCAT) deficiency. Description of two new mutations in the LCAT gene. Nephrol Dial Transplant. 2004;19:1622-4.
- [Google Scholar]
- Histopathology of corneal changes in lecithin-cholesterol acyltransferase deficiency. Cornea. 2002;21:834-7.
- [Google Scholar]
- Renal transplantation in patients with familial lecithin: Cholesterol-acetyltransferase deficiency. Transplant Proc. 1977;9:1665-71.
- [Google Scholar]
- Long-term follow-up of a patient with lecithin cholesterol acyltransferase deficiency syndrome after kidney transplantation. Transplantation. 1993;56:233-6.
- [Google Scholar]




