

Translate this page into:
Dorfman–Chanarin Syndrome with Renal Involvement: A Rare Case Report and Literature Review
Address for correspondence: Dr. Ikram Agrebi, Department of Nephrology, Hedi Chaker University Hospital, Ain Road Km 0.5, Sfax, Tunisia. E-mail: ikram_agrebi@yahoo.fr
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Dorfman–Chanarin syndrome (DCS) is a rare autosomal recessive disease. It is a multisystemic disease in which renal involvement is uncommon. We report the case of a woman with nephrotic syndrome associated with DCS. A 36-year-old woman was referred to the nephrology department for edema with known history for DCS. On physical examination, she had ichthyosiform erythroderma with generalized scaly skinand ascites. The ophthalmologic examination revealed a cataract in the right eye. Abdominal ultrasound examination showed hepatomegaly and splenomegaly. Laboratory tests showed normal renal and liver function. The blood cell count showed pancytopenia. Immunologic exams showed the presence of anti-mitochondrial antibodies. Kidney biopsy showed mesangial proliferative glomerulonephritis with extensive lipid vacuoles in the tubular epithelial cells. Immunofluorescence study showed mesangial deposits of IgG, C3, kappa, and lambda. To the best of our knowledge, this is the first case of DCS with renal involvement reported in an adult.
Keywords
Dorfman–Chanarin syndrome
kidney biopsy
nephrotic syndrome

Introduction
Dorfman–Chanarin syndrome (DCS) is a rare autosomal recessive disease. It is caused due to a mutation in the ABHD5 gene, which codes for proteins that enable the activation of adipose triglyceride lipase (ATGL) and leads to the accumulation of lipid vacuoles in multiple types of cells. The main clinical feature is the non-bullous congenital ichthyosiform erythroderma (NBCIE). It is a multisystemic disease. Renal involvement is uncommon. We report the case of a woman with nephrotic syndrome associated with DCS.
Case Report
A 36-year-old woman was admitted to the nephrology department for edema. Her past medical history was notable for DCS, which was diagnosed in the dermatology department. She was a product of nonconsanguineous marriage. She was initially referred to the gastroenterology department for hepatic steatosis, ascites, and cirrhosis. FibroScan confirmed advanced liver scarring with a value of 46.4 kPa. Esogastroduodenal endoscopy was normal. Laboratory findings indicated a urinary protein excretion of 16 g/day with serum albumin of 1.8 g/dL. On admission in our department, her physical examination showed a height of 175 cm and a weight of 67 kg. Her blood pressure was 120/71 mmHg. She had ichthyosiform erythroderma with generalized scaly skin. Also, she had ascites and lower limb edema. Cardiovascular and neurologic examinations were normal. She had no muscular weakness. The ophthalmologic examination revealed a cataract in the right eye. The otologic examination showed no signs of deafness.
Laboratory tests showed normal renal and liver function with normal levels of alkaline phosphatase and γ-glutamyltransferase. The blood cell count showed pancytopenia with a hemoglobin level of 6.4 g/dL and platelet number of 83,000/mm3. Nephrotic syndrome was confirmed with 7.7 g/day of proteinuria and low serum albumin level at 18 g/L. Immunologic exams showed the presence of anti-mitochondrial antibodies. Serological tests for hepatitis B, C, and human immunodeficiency virus (HIV) were negative. Phospholipase A2 receptor (PLA2R) antibodies were negative. Accessory salivary gland biopsy showed no amyloid deposits. A kidney biopsy was performed after platelet transfusion. It showed mesangial proliferative glomerulonephritis with extensive lipid vacuoles in the tubular epithelial cells [Figure 1]. Immunofluorescence study showed mesangial deposits of IgG, C3, kappa, and lambda [Figure 2]. Abdominal ultrasound examination showed hepatomegaly with rough liver edges, enlarged portal vein, splenomegaly, and ascites.

- Kidney biopsy findings of a patient with Dorfman–Chanarin syndrome. (a) Light microscopy demonstrates the presence of mesangial proliferation without crescents or sclerosis (trichrome stain). (b) Presence of vacuoles in the tubular epithelium of kidneys (trichrome stain). (c) Oil red O staining demonstrates the accumulation of lipid droplets in the tubular vacuoles
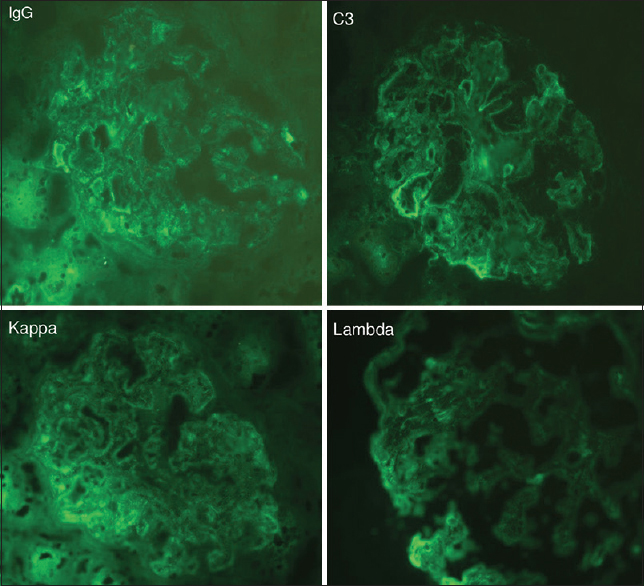
- Immunofluorescence findings: mesangial immune deposits of IgG, C3, kappa, and lambda
Discussion
DCS is an extremely rare metabolic disease.[1] It is an autosomal recessive inherited disease that leads to a lipid storage disorder. In 1953, Jordan identified lipid vacuoles in the cytoplasm of leukocytes from the peripheral blood smear of two brothers who suffered from progressive muscular dystrophy.[2] The first cases were described by Dorfman et al. in 1974[3] and Chanarin et al. in 1975.[4] They reported an abnormality of triglyceride storage with the presence of lipid vacuoles in leukocytes associated with various clinical signs, essentially cutaneous but also neurologic, ophthalmologic, otologic, and hepatic.
To date, nearly 150 cases of DCS have been reported in the literature. The median age of presentation is 9 years. Most of the cases are from the Mediterranean basin and the Middle East. Turkey is the country where the syndrome is the most observed, followed by India.[5] DCS is caused by a mutation in the ABHD5 gene located on the short arm of chromosome 3 that codes for factors helping the ATGL enzyme in the release of fatty acids from the intracellular triacylglycerol stores of different tissues. This disrupts intracellular lipolysis and induces tissue lipid overload.
DCS is a multisystemic disease. The two major characteristic signs are skin manifestations and the presence of lipid vacuoles in the cytoplasm of various cells. Skin involvement is characterized by an NBCIE.[6,7] Other organs may be affected. Liver involvement is common and may be present in 86% of cases.[5] The most frequent manifestation is liver steatosis, which may progress to cirrhosis, as in our patient.[8] Myopathy with slowly progressive weakness of muscle is not exceptional. This symptom was first described by Jordan in 1953.[2] This sign could be found in 59% of patients.[5]
Ocular manifestations may occur in the form of cataracts, strabismus, nystagmus, myopia, and ectropion.[9] Neurologic involvement may include mental retardation, ataxia, microcephaly, and neurosensory deafness.[5] Manifestations may vary according to ethnicity and mutation type.[10]
Renal involvement in DCS is rare. It has been reported in only three case reports [Table 1]. The first one was published in 2002.[11] It reported the case of a 5-year-old girl who presented with nephrotic syndrome, ascites, and splenomegaly. She was started on prednisolone given as a single dose of 60 mg/m²/day for 8 weeks. She showed a partial response to the treatment. A kidney biopsy was performed, which showed mesangial proliferation. She was given methylprednisolone pulse therapy for three consecutive days, with the addition of cyclophosphamide at a dose of 2 mg/kg/day for 3 months, which led to remission.
| Age at DCS presentation | Age of nephropathy onset | Dorfman symptoms | Proteinuria | Creatinine | Kidney biopsy findings | |
|---|---|---|---|---|---|---|
| Case 1[11] | Congenital onset | 5 years | •Short stature | 400 mg/m² | Normal | Mesangial proliferative glomerulonephritis |
| •Congenital ichthyosiform | ||||||
| erythroderma | ||||||
| •Generalized body puffiness | ||||||
| •Ascites | ||||||
| •Hepatomegaly | ||||||
| •Hypoalbuminemia | ||||||
| •Dyslipidemia | ||||||
| Case 2[12] | 3 weeks | 8 months | •Short stature | Positive | Initially normal, then occurence of renal failure | Minimal proliferation of podocytes. Mesangial expansion. Segmental sclerosis in one out of 40 glomeruli. Tubulointerstitial elements were normal. Focal lipid vacuoles detected in the distal and collecting tubular epithelium |
| •Congenital ichthyosiform | ||||||
| erythroderma | ||||||
| •Hepatosplenomegaly | ||||||
| •Neurologic developmental delay | ||||||
| •Jordan anomaly fatty liver | ||||||
| •Muscle involvement | ||||||
| •Positive anti-mitochondrial antibody | ||||||
| •Hypoalbuminemia | ||||||
| •Hypergammaglobulinemia | ||||||
| •Hypereosinophilia | ||||||
| •Elevated immunoglobulin E | ||||||
| Case 3[13] | Third day of birth | 17 years | •Coarse facies | Subnephrotic range proteinuria | Marginally raised (2.4 mg/dL) | Extensive lipid vacuoles in the tubular epithelial cells |
| •Short stature | ||||||
| •Hepatosteatosis | ||||||
| •Sensorineural deafness | ||||||
| •Mild mental retardation | ||||||
| •Early cataract | ||||||
| •Edema | ||||||
| •Elevated liver enzymes | ||||||
| Our case | Congenital onset | 36 years | •Ichthyosiform erythroderma | 7.7 g/day | Normal | Mesangial proliferative glomerulonephritis with lipid vacuoles in the tubular epithelial cells |
| •Cataract | ||||||
| •Hepatosplenomegaly | ||||||
| •Ascites | ||||||
| •Lower limb edema | ||||||
| •Hypoalbuminemia |
The second case was a 7-month-old girl with a history of recurrent respiratory infections and NBCIE.[12] She died at the age of 8 months from severe renal failure. The kidney biopsy showed mesangial proliferation, segmental sclerosis in one out of 40 glomeruli, and lipid vacuoles detected in the collecting tubular epithelium. The third case was a 17-year-old Indian child who presented with nephritic syndrome.[13] Renal biopsy showed lipid vacuoles in the tubular epithelium. He was given 0.5 mg/kg/day of prednisolone in a tapering dose and diuretics. His kidney function was normal, except for persistent proteinuria.
In a recently published article,[14] four of nine patients who underwent abdominal ultrasound showed abnormal kidney. Three patients had poorly differentiated kidney parenchyma and one patient had an enlarged kidney. This may show that the kidney could be a more commonly involved organ than previously reported in the literature.
Conclusion
To the best of our knowledge, this is the first case of DCS with renal involvement reported in an adult. The association between the two was confirmed by the presence of lipid vacuoles in the tubular epithelium. The nephrotic syndrome presentation in our patient was similar to the first case previously described. Biopsy results showed commonalities in these cases. Although there is no well-defined protocol for treating renal damage, it seems to be effective in mitigating proteinuria and raising plasma albumin levels.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Truncation of CGI-58 Protein causes malformation of lamellar granules resulting in ichthyosis in Dorfman-Chanarin syndrome. J Invest Dermatol. 2003;121:1029-34.
- [Google Scholar]
- The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva (ERB) Acta Med Scand. 1953;145:419-23.
- [Google Scholar]
- Neutral-lipid storage disease:A new disorder of lipid metabolism. Br Med J. 1975;1:553-5.
- [Google Scholar]
- Erythrokeratoderma variabilis-like ichthyosis in Chanarin-Dorfman syndrome. Br J Dermatol. 2005;153:838-41.
- [Google Scholar]
- Clinical and genetic characterization of Chanarin-Dorfman syndrome. Biochem Biophys Res Commun. 2008;369:1125-8.
- [Google Scholar]
- Chanarin–Dorfman syndrome in three siblings in a non-consanguineous family. J Eur Acad Dermatol Venereol. 2016;30:157-9.
- [Google Scholar]
- Chanarin-Dorfman syndrome:Genotype-phenotype correlation. Eur J Med Genet. 2015;58:238-42.
- [Google Scholar]
- Dorfman-Chanarin syndrome (neutral lipid storage disease):New clinical features. Br J Dermatol. 2001;144:430-2.
- [Google Scholar]
- Renal involvement as a rare complication of Dorfman–Chanarin syndrome:A case report. Pediatr Dermatol. 2008;25:326-31.
- [Google Scholar]
- Chanarin–Dorfman syndrome with rare renal involvement. Br J Dermatol. 2017;176:545-8.
- [Google Scholar]
- Thyroid involvement in Chanarin-Dorfman syndrome in adults in the largest series of patients carrying the same founder mutation in ABHD5 gene. Orphanet J Rare Dis. 2019;14:112.
- [Google Scholar]




