

Translate this page into:
Hypercalcemia in an Infant with Primary Hyperoxaluria Type 2: A Novel Association
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Hypercalcemia in infants presents with a variety of clinical features and the etiology of hypercalcemia varies with age. Here we present a case of hypercalcemia in an infant presenting with nephrocalcinosis and nephrolithiasis. Our investigations led us to a diagnosis of primary hyperoxaluria (PH) type 2, a rare metabolic disorder, along with hypercalcemia, a never before reported association. A 9-month-old female presented with urinary tract infection and systemic features requiring hospitalization and parenteral antibiotics. Investigations revealed bilateral medullary nephrocalcinosis. Genetic testing revealed a diagnosis of Primary hyperoxaluria type 2 with two possible mutations. Sanger sequencing of the parents identified the pathogenic mutation in the mother. This is the first report of a genetically proven case of primary hyperoxaluria type 2 associated with hypercalcemia.
Keywords
Hypercalcemia
nephrocalcinosis
next generation sequencing
primary hyperoxaluria type 2

Introduction
Hypercalcemia in infancy is less common compared to adults.[1] Moreover, normal serum calcium concentrations are higher in children than in adults, and age-specific cut-offs are required to make a diagnosis.[2] Clinical features of hypercalcemia in infants can range from non-specific manifestations to recurrent urinary tract infection (UTI) due to nephrolithiasis with failure to thrive and neurological manifestations like seizures and hypotonia.[13]
Primary hyperoxaluria (PH) is a rare cause of nephrocalcinosis and end-stage renal disease. Three types of PH have been described to date depending on enzyme defects in the oxalate metabolic pathway. PH type 1 (PH1) is caused by the low or absent activity of liver-specific peroxisomal alanine: glyoxylate aminotransferase (AGT), which results in increased urinary excretion of both oxalate and glyoxalate.[4] In PH type 2 (PH2), deficiency or absence of glyoxylate reductase/hydroxy-pyruvate reductase (GRHPR) leads to elevated urinary excretion of both oxalate and L-glyceric acid.[5] The third type of PH (PH3) was recently found to be due to an abnormality of the enzyme 4-hydroxy-2-oxaloglutarate aldolase (HOGA1). Though the classical manifestation of PH does not include hypercalcemia, some cases of hypercalcemia have been reported with PH1. The mechanism of hypercalcemia in these cases is non-PTH dependent and may involve 1,25 dihydroxy cholecalciferol or parathormone related peptide (PTHrP).[6] As PH2 is an extremely rare condition, no prevalence data exist. It is thought to be less common than primary hyperoxaluria type 1, which has a prevalence of approximately 1:1,000,000.[7] Here we present a case of hypercalcemia in PH2, a novel association.
Case Presentation
A female infant (1st born child) aged 8.5 months, born out of non-consanguineous marriage, presented with fever for 2 weeks. Fever was intermittent & high grade. She was hospitalized and evaluated for the same, with investigations showing urinary tract infection (UTI). The infection resolved after parenteral meropenem & amikacin administration. As a part of the workup for UTI, ultrasonography of the abdomen was asked which showed bilateral medullary nephrocalcinosis. Serum biochemistry showed an elevated calcium level which was confirmed on multiple testing. The hypercalcemia persisted even after resolution of the UTI. There was no history of seizures, loss of consciousness, graveluria or hematuria. There was no history of intake of non-dietary calcium either oral or intravenous. The infant was exclusively breastfed for the first 6 months of her life. There was no history of any fractures or bony abnormalities. There was no history of renal disease or recurrent UTI in the family. At birth, she weighed 3 kgs, had an unremarkable perinatal history and the developmental milestones were satisfactory. The clinical examination of the infant did not provide additional information and we decided to proceed with investigations. Results of complete urinalysis and metabolic evaluation are shown in Table 1.
| Parameters | Results | Reference Range | |
|---|---|---|---|
| At presentation | 6 months follow-up | ||
| Blood urea | 16 mg/dl | 5-18 | |
| Serum creatinine | 0.24 mg/dl | 0.25 | 0.2-0.4 |
| Estimated GFR (Schwartz formula) | 111 ml/min/1.73 m2 | 110 ml/min/1.73 m2 | |
| Serum ionized calcium | 2 mmol/L | 1.9 | 1.2-1.38 |
| Serum total calcium (mean) | 11.6 mg/dl | 11.4 | 8.7-11 |
| Serum Albumin | 3.9 g/dl | 4.0 | 3.5-4.5 |
| Serum phosphorus | 4.7 mg/dl | 3.8-6.5 | |
| Serum PTH (ECLIA) | 4.35 pg/ml | 15-65 | |
| Serum 1,25 (OH) 2 Vitamin D3(CLIA) | 29.4 pg/ml | 19.9-79.3 | |
| Serum Alkaline Phosphatase | 184 IU/L | 124-341 | |
| Serum 25 (OH) Vit D | 34 ng/ml | 30-100 | |
| fT4 | 1 ng/dl | 0.9-2.5 | |
| TSH | 1.6 mIU/ml | 0.5-4.5 | |
| Spot urine calcium/creatine | 0.45 mg/ | 0.2 | <0.6 mg/mg |
| Urine Examination | |||
| Urinalysis pH | 6.2 | 6.3 | 4.6-8.0 |
| Specific gravity | 1.010 | 1.016 | |
| RBC | Absent | Absent | |
| Pus cells | 3/HPF | 1-2/HPF | |
| Casts or crystals | Oxalate crystals | Oxalate crystals | |
| C/S | E. coli >105 CFU/ml | No growth | |
| ABG | |||
| pH | 7.41 | 7.35-7.45 | |
| Bicarbonate | 20.5 mEq/L | 20-24 | |
| pCO2 | 30 mmHg | ||
Ultrasonography (USG) of kidney, ureter and urinary bladder showed bilateral increased medullary echogenicity involving renal pyramids with sparing of cortex suggestive of bilateral medullary nephrocalcinosis along with tiny nephrolithiasis [Figure 1a]. Repeated measurements of albumin adjusted total serum calcium yielded values higher than age specific cut-offs. (all serum calcium blood samples were obtained without using a tourniquet The investigations were suggestive of Parathormone (PTH) independent hypercalcemia. The examination of urine revealed transparent bipyramidal or envelope-shaped oxalate crystals [Figure 1b].
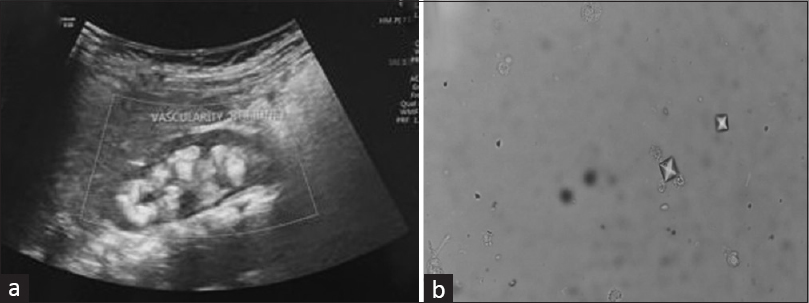
- (a): USG kidney showing medullary nephrocalcinosis; (b): Urine showing bipyramidal oxalate crystals
Having failed to ascertain a cause for this hypercalcemia, genetic testing was planned and the genetics laboratory conducted the following analysis. DNA was extracted from the index patient and whole exome sequencing was done. All 22, 000 genes were analysed. The genes analysed list and coverage is attached as an additional file (supplementary file).
Sequencing of the protein coding regions of approximately 30 Mb of the human exome [targeting approximately 99% of regions in consensus coding sequence project (CCDS) and RefSeq)] was performed using Illumina next generation sequencing (NGS) systems at a mean depth of 50-60X and % of bases covered at 20X depth >90% in the target region. In some cases, due to the complexity of the sequence, not all variants in the flanking regions could be analyzed. Genome analysis toolkit (GATK) best practice framework was followed for variant identification. Burrows Wheeler aligner (BWA-mem aligner) was used to align the obtain sequences to human reference genome (GRCh37/hg19). Duplicate reads identification and removal, base quality recalibration and re-alignment of reads based on indels were done using inbuilt Sentieon modules. Sention’s Haplotypecaller module has been used to identify the variants which are relevant to the clinical indications. Along with this Deep variant analysis pipeline on Google cloud platform was used as a secondary pipeline to call genetic variants. Quality checks (QC) were performed on all variant cell format (VCF) files to exclude variants where sequencing is of poor quality. Additional QC metrics includes total homozygous and heterozygous calls (Single Nucleotide variants and indels), proportion of variant calls that were common, number of variants falling into different annotated consequence categories, number of extreme heterozygotes (alternate allele proportion 0.8). Variant annotations were done using published databases like OMIM, GWAS, GNOMAD, 1000Genome etc. Non-synonymous and splice site variants were used for clinical interpretation. Silent variations that do not result in any change in amino acid in the coding region are not reported.
Sequencing of exons and the intron-exon boundaries in the GRHPR gene (NM_012203.2) revealed a compound heterozygous state of two nucleotide variants. Heterozygous variant c. 494G>A (p. Gly165Asp) was found in exon 6. In exon 8, we identified mutation c. 800G>A in heterozygous form. The former mutation is pathogenic for PH2, whereas the latter was a variance of uncertain significance for the same condition. The missense variant NM_012203.2 (GRHPR): c. 800G>A (p. Gly267Glu) has not been reported previously as a pathogenic variant nor as a benign variant, to our knowledge. The p. Gly267Glu variant is novel (not in any individuals) in gnomAD. The p. Gly267Glu variant is novel (not in any individuals) in 1 kG. There is a moderate physicochemical difference between glycine and glutamic acid. The p. Gly267Glu missense variant is predicted to be damaging by both SIFT and PolyPhen2. The glycine residue at codon 267 of GRHPR is conserved in all mammalian species. The nucleotide c. 800 in GRHPR is predicted conserved by GERP++ and PhyloP across 100 vertebrates.
Segregation study in parents for the point mutation were done by Sanger sequencing. Targeted sequencing and mutation analysis was performed by Polymerase Chain Reaction (PCR) followed by automated DNA sequencing of the amplicon using BigDye ABI Genetic Analyzer 3500DX platform. The identified variants were at depth at 102 and 132 respectively. The classification was done according to the American College of Medical Genetics and Genomics (ACMG) criteria.[8]
Sanger sequencing of the parents’ DNA revealed the pathogenic mutation in the mother and the latter mutation in the father [Figures 2 and 3]. Serum biochemistry and ultrasound kidney for both parents were obtained following the genetics reports. All reports in both parents were essentially normal including serum calcium.
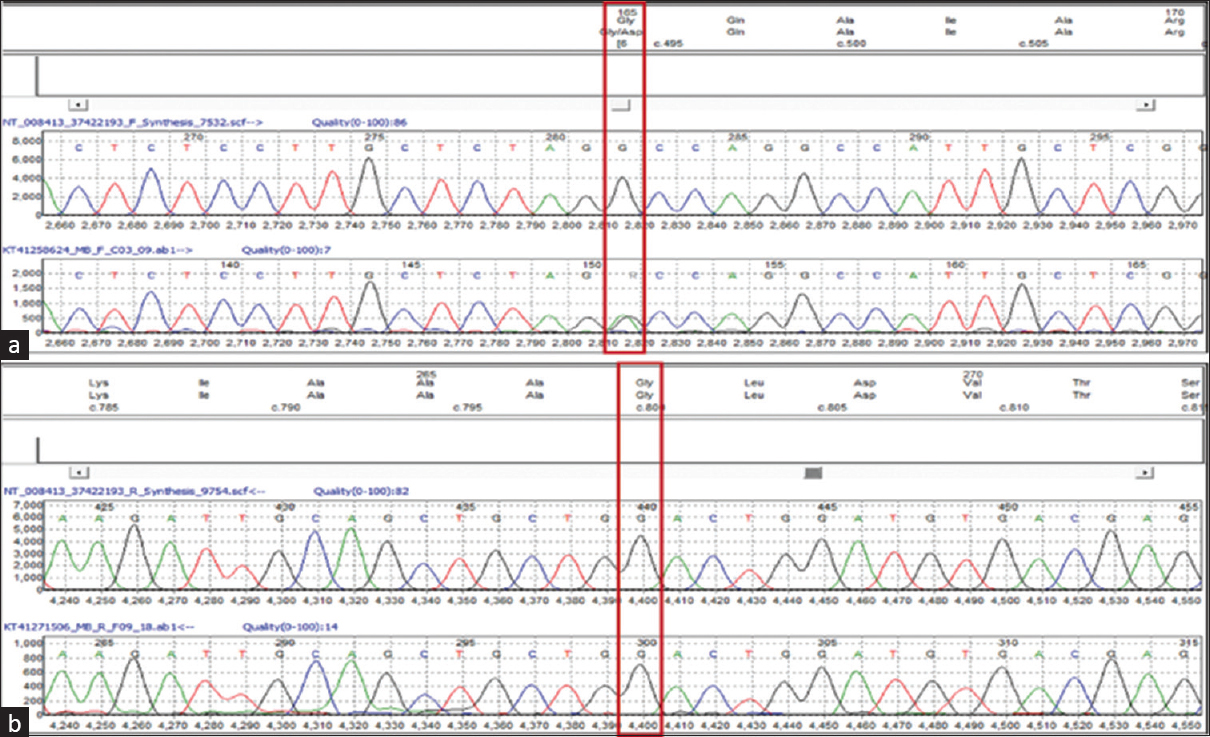
- Sanger sequencing data (electropherogram) for the mother sample showing nucleotide change at chr9: c. 494G > A, (p. Gly165Asp) in GRHPR gene (a) and no nucleotide change at chr9: c. 800G > A, (p. Gly267Glu) in GRHPR gene (b)

- Sanger sequencing data (electropherogram) for the father sample showing nucleotide change at chr9: c. 800G > A, (p. Gly267Glu) in GRHPR gene (a) and no nucleotide change at chr9: c. 494G > A, (p. Gly165Asp) in GRHPR gene (b)
The infant was managed for UTI and she responded to the antibiotics. Adequate fluid intake was advised and keeping in view the diagnosis of PH2, urinary alkalizer therapy was initiated. The dose of alkali was 5 meq/kg/day. A low oxalate diet has also been advised. The parents were advised to avoid diet rich in potato, spinach, chocolates and soy products while feeding the child. Diet rich in bananas, papaya and milk products were encouraged. The patient was discharged in afebrile, stable condition.
The infant is doing well at follow up, she is gaining weight, there have been no further episodes of fever related to urinary infection. Investigations done at six months follow up showed persistent hypercalcemia in the absence of any evidence of UTI without deterioration of renal function.
Discussion
Nephrolithiasis and nephrocalcinosis are uncommon in infants. Our case is one such with an initial presentation of nephrocalcinosis and urinary tract infection. This is similar to most cases of nephrocalcinosis in infants and toddlers in whom the first clinical symptom, if any, are gross or microscopic haematuria and/or leukocyturia which may be misdiagnosed as UTI or cystitis. Nevertheless, nephrocalcinosis warrants thorough evaluation for determining a cause and avoid or slow renal damage.[9]
In our case, detailed biochemistry including spot urine analyses for nephrocalcinosis did not yield significant results. Blood biochemistry and urine for calcium, creatinine, citrate, oxalate, phosphate etc., should be done for all patients of nephrocalcinosis preferably in a properly collected 24 hr urine sample. However, in infants or young children, or in situations where a 24 h urine collection is difficult, random urine measurements, using the ratio of the concentration of each analyte to that of urine creatinine, provide valuable information.[10]
Primary hyperoxaluria type 2 was ascertained as the cause for nephrocalcinosis in our patient based on findings of whole genome sequencing in the index case and the pathogenic variant was traced to the mother by sanger sequencing of both parents. Primary hyperoxaluria is a recognised cause of early nephrocalcinosis, nephrolithiasis and end stage renal disease (ESRD). The most common type, PH1 has been associated with renal stones by imaging in up to 60% and nephrocalcinosis in 40%.[11] PH2 which is an extremely rare entity, is mostly diagnosed in childhood and presents with hematuria, renal colic and recurrent urinary tract infection and obstruction. The majority of individuals have renal stones composed of calcium oxalate. Nephrocalcinosis, observed on ultrasound examination, abdominal x-ray, or CT examination, is a much less common finding in PH2 than in PH1. The disease can progress to ESRD although this outcome appears to be later in PH2 than in PH1, in which 50% of affected individuals develop ESRD by age 25 years.[12]
Hypercalcemia is another important finding in our case. Our workup did not reveal any cause of hypercalcemia typically found in this age group and the hypercalcemia persisted even after UTI had resolved and at six months follow-up. Some cases of hypercalcemia have been reported in PH1 but there is no mention of hypercalcemia in PH2 in literature. One case series of hyperoxaluria patients has a mention of hypercalcemia in PH2, but that was discovered four years after dialysis was initiated in the subject.[13] Renal function in our child was normal when hypercalcemia was first detected and the renal function was normal even at six months follow-up.
To conclude, to the best of our knowledge this is the first ever case report of hypercalcemia associated with PH2. Whereas PH2 itself is extremely rare and very few cases have been reported in literature, this presentation with hypercalcemia is novel. The cause of hypercalcemia in this case could not be delineated by our workup and more research may through light on this association
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Calcium-regulating hormones and minerals from birth to 18 months of age: A cross-sectional study. II. Effects of sex, race, age, season, and diet on serum minerals, parathyroid hormone, and calcitonin. Pediatrics. 1986;77:891-6.
- [Google Scholar]
- Investigation and management of hypercalcaemia in children. Arch Dis Child. 2012;97:533-8.
- [Google Scholar]
- Primary hyperoxaluria:from gene defects to designer drugs? Nephrol Dial Transplant. 2005;20:1525-9.
- [Google Scholar]
- Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat. 2003;22:497.
- [Google Scholar]
- Hypercalcaemia complicating systemic oxalosis in primary hyperoxaluria type 1. Nephrol Dial Transplant. 1995;10((Suppl 8)):17-21.
- [Google Scholar]
- Primary hyperoxaluria type 2. 2008 Dec 2 [Updated on 2017 Dec 21] In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2021.
- [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
- [Google Scholar]
- Diagnostic examination of the child with urolithiasis or nephrocalcinosis. Pediatr Nephrol. 2010;25:403-13.
- [Google Scholar]
- Nephrocalcinosis is a risk factor for kidney failure in primary hyperoxaluria. Kidney Int. 2015;87:623-31.
- [Google Scholar]
- Granulomatous inflammation and hypercalcemia in patients with severe systemic oxalosis. Kidney Int Rep. 2021;7:343-9.
- [Google Scholar]




