

Translate this page into:
Fabry Disease: Report of Two Cases with Uncommon Presentation
Address for correspondence: Dr. Pallavi Prasad, Department of Pathology, Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow - 226 014, Uttar Pradesh, India. E-mail: pallavisgpgi@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Fabry disease (FD) is a rare, lysosomal storage disorder characterized by multiorgan accumulation of predominantly globotriaosylceramide (GL3) and its metabolite. Resulting renal, cardiac, and cerebrovascular complications are crucial causes of morbidity and mortality in FD. Enzyme replacement therapy (ERT) shows promising outcomes for these patients, provided that therapy is initiated early. Thus, precise and early diagnosis of the disease is a pivotal factor determining the corollary of the disease. We report two cases of young adult males who presented to the nephrology department with proteinuria. A kidney biopsy was performed in both cases, which was suggestive of FD. The final conclusive diagnosis of FD was provided by electron microscopy.
Keywords
Electron microscopy
Fabry disease
myelin figures
proteinuria
renal biopsy
white urine

Introduction
Fabry disease (FD) is an X-linked inherited disorder of glycosphingolipid metabolism with an estimated birth prevalence of 1:40,000–117,000.[1] It is caused due to absent or reduced activity of lysosomal exoglycohydrolase, α-galactosidase A. There is deposition of globotriaosylceramide (GL3) within lysosomes in several cell types such as renal, cardiac, endothelial, cardiac, and dorsal root ganglion neuronal cells. This leads to characteristic features such as albuminuria, acroparesthesia, anhidrosis, disseminated angiokeratoma, cornea verticillata, and gastrointestinal symptoms.
In kidneys, the progressive accumulation of GL3 may lead to chronic kidney disease (CKD), which is the main cause of premature death in the classical phenotype. Impaired glomerular filtration rate (GFR), proteinuria, and tubular dysfunction are the main manifestations of renal disease.[2] These are usually seen to develop during the third decade of life.[345] The classically affected males may then progress to end-stage renal disease (ESRD), usually early in the fifth decade. However, in females, the rate of progression to ESRD or transplantation is much slower.[6]
Case Reports
Case 1
A 23-year-old male with no known comorbidities presented to the nephrology department of our institute with complaints of intermittent episodes of passage of milky white urine, edematous illness for the past 7 years, dysuria, hesitancy, incomplete voiding of urine for the past 3 years, and off-and-on fever. He had one episode of gross hematuria 2 years back. The patient gave a history of filariasis being endemic at his native place. He gave no history of rash, oliguria, allergies, or any drug intake.
On examination, his blood pressure was 130/80 mm Hg, pulse rate was 80/min, and edema was present. The rest of the systemic examination was within normal limits. He underwent workup for lower urinary tract symptoms and milky white urine. Urine routine examination showed mild proteinuria (1+), nil RBCs, and 3–4 WBCs/HPF. Urine fat/chyle was positive. There was no hypercalciuria or hyperphosphaturia. 24-h urine protein was 2.08 g, and serum creatinine was 0.8 gm/dL. Lipid profile was normal (triglyceride: 152 mg/dL, HDL cholesterol: 40 mg/dL, VLDL cholesterol: 30.4 mg/dL, and total cholesterol: 130.6 mg/dL). The hematological parameters were within normal limits (Hb: 13.1 gm/dL, TLC: 8.3 × 1000/uL, platelet count: 226 × 1000/mm3). There was no peripheral eosinophilia (N65 L30 E2 M3). He also underwent workup for genitourinary tuberculosis in view of lower urinary tract symptoms. Urine microscopy and cultures for bacteria, fungal elements, and mycobacteria were negative.
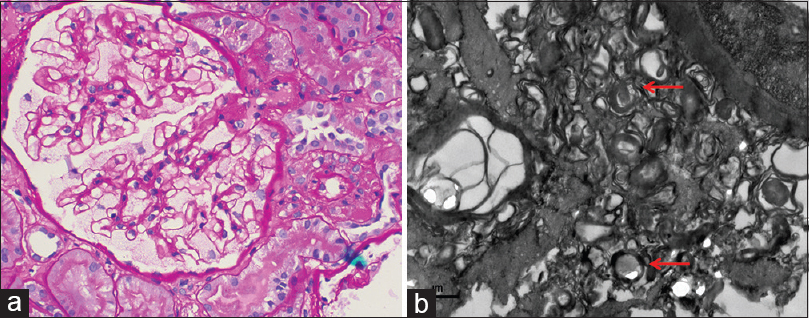
The clinical differential diagnoses considered in view of subnephrotic proteinuria, normal serum creatinine, and bland sediments were focal segmental glomerulosclerosis and IgA nephropathy. In addition, empirically, the patient was started on diethylcarbamazine (DEC), and a kidney biopsy was performed. It comprised a total of six glomeruli (PAS), of which one was globally sclerosed. The remaining five viable glomeruli were enlarged and showed distended hypertrophic glomerular visceral epithelial cells (podocytes) with foamy vacuolated appearance [Figure 1a and 1b]. Tubules showed minimal atrophy. Interstitium showed minimal fibrosis and minimal lymphomononuclear infiltrate, including aggregates of foam cells. Blood vessels were largely unremarkable. No microfilarial parasite was identified. Direct immunofluorescence showed 13 glomeruli with no immune deposits.

- Renal biopsy from thefirst case containing six glomeruli, all showing prominent vacuolization of the podocytes (a) (PAS, ×10). High-power view of single enlarged glomerulus displaying foamy-appearance (b) (PAS × 40). Electron micrograph showing numerous electron-dense lamellated bodies (myelin figures or zebra bodies) in the cytoplasm of visceral epithelial cells (red arrow) and endothelial cells (green arrow) (c) (uranyl acetate and lead citrate). Myelin figures in the peritubular capillaries (red arrow) and tubular epithelial cells (green arrow) (d) (uranyl acetate and lead citrate)
Case 2
A 24-year-old male presented to the nephrology department for evaluation of sub-nephrotic range proteinuria with bland sediments. He had edematous illness 11 years back for which he was on a suboptimal dose of steroids. He gave no history of hematuria, dysuria, fever, rash, oliguria, allergies, or any drug intake. On examination, he was edematous and anemic, with a blood pressure of 140/80 mm Hg. The rest of the systemic examination was within normal limits.
Urine routine examination showed moderate proteinuria (2+) and nil RBCs and WBCs. 24-h urine protein was 1.41 g, and serum creatinine was 1.3 gm/dL. Lipid profile was normal (triglyceride: 65 mg/dL, HDL cholesterol: 34 mg/dL, VLDL cholesterol: 13.0 mg/dL, and total cholesterol: 181 mg/dL). The hematological parameters were within normal limits (Hb: 10.1 gm/dL, TLC: 6.45 × 1000/uL, platelet count: 161 × 1000/mm3). There was no peripheral eosinophilia (N45 L342 E6 M7).
The clinical differential diagnoses considered were focal segmental glomerulosclerosis and IgA nephropathy. A kidney biopsy was performed. It comprised a total of 15 glomeruli, of which four were globally sclerosed and one showed segmental sclerosis. The unsclerosed glomeruli were enlarged with mild mesangial widening. Glomerular visceral epithelial cells were distended, displaying a foamy appearance with numerous fine cytoplasmic vacuoles [Figure 2a]. Similar vacuolation was also observed in distal tubular epithelial cells and renal interstitium. Patchy mild tubular atrophy and interstitial fibrosis were also noted. Blood vessels showed mild intimal fibrosis. Direct immunofluorescence showed 10 glomeruli displaying coarse granular C3 (3+) deposits along the glomerular capillary wall and in the mesangium. No immune deposits for IgG, IgM, IgA, and C1q were noted.
- High-power view of renal biopsy from second case showing single enlarged glomerulus displaying foamy-appearance (a) (PAS × 40). Electron micrograph showing electron-dense lamellated bodies (myelin figures or zebra bodies) in the cytoplasm of visceral epithelial cells (red arrow) (b) (uranyl acetate and lead citrate)
In both cases, the histological differential diagnosis considered in view of the foamy appearance of the glomeruli were storage disorders including Fabry's disease, LCAT deficiency, crystal-storing histiocytosis, histiocytic glomerulopathy, thrombotic microangiopathy, and lipoprotein glomerulopathy. Electron microscopy was subsequently performed in both cases. It revealed the presence of numerous myelin figures (or zebra bodies) composed of electron-dense, multilamellated concentric layers, having a periodicity of 3.5–5 nm inside the podocytes, distal tubular epithelial cells, and endothelial cells of the glomerular capillary loops, as well as peritubular capillaries [Figure 1c, 1d, and 2b]. No crystals, histiocytic foam cells, features suggestive of thrombotic microangiopathy, lipid material were evident. Thus, the final diagnosis of FD was rendered.
Following the histological diagnosis, both patients were reevaluated for features of FD. Case 1 gave a history of acroparaesthesia in the form of tingling, prickling, and feeling of burning in the hands and feet, whereas case 2 was found to have skin lesions in the form of small, dark, red spots. The diagnosis was further confirmed by alpha-galactosidase-A level enzyme estimation, which came out to be 5.54 nmol/h/mg (normal subjects: 21–175 nmol/h/mg), mean = 65 nmol/h/mg and GLA gene-g. 9356_9357 del CA (NM_000169) pathogenic mutation in case 2.
Discussion
FD is an X-linked lysosomal storage disorder caused by the mutation of enzyme α-galactosidase A (α-GAL), resulting in the accumulation of glycolipids, predominantly GL3, and its metabolite (lyso-GL3), in multiple organs. It affects both the sexes. The “classical phenotype” with an enzyme activity of <1% is found in males and is characterized by early-onset of disease manifestation. The “late-onset phenotype” is seen in males with an enzyme activity of >1%, as well as in females, who may even have normal levels of enzyme activity. Initial manifestations include dermal and gastrointestinal symptoms. As the accumulation of glycolipids continues, corneal, auditory, and cognitive defects ensue. With further increasing age and continuous accumulation, cardiac, renal, and cerebrovascular manifestations appear, which can lead to catastrophic events.
Fabry nephropathy (FN), that is, renal involvement, is reported to have a prevalence of 0.20–0.99% in male and 0.05–0.33% in female patients on dialysis.[7] Progressive glycolipid accumulation, inflammatory mediators, oxidative stress, overexpression of adhesion molecules, increased cytokines such as TGF-β, apoptosis, decreased mTOR and protein kinase B signaling pathways, and angiotensin-II have been implicated in the pathogenesis of FN.[7] Clinical findings include albuminuria, proteinuria, progressive decline in glomerular infiltration rate (GFR), and presence of renal cysts, mainly parapelvic cysts. GL3 accumulates in all the renal cells, of which podocytes, endothelial cells, and mesangial cells play the most important role in disease progression. As the disease progresses, podocytes and endothelial cells become hypertrophic, with foamy vacuoles. Later in the disease course, irreversible changes in the form of glomerulosclerosis, tubular atrophy, interstitial fibrosis, and arterial and arteriolar sclerosis occur. Thus, a progressive transition occurs from the early microalbuminuria and proteinuria in the second decade of life to the chronic kidney disease (CKD), requiring renal replacement therapy between the fourth and fifth decade of life.
Renal biopsy is the most confirmatory tool for the diagnosis of FN irrespective of the level of proteinuria and renal dysfunction because it determines the amount of irreversible damage. Moreover, it plays an important role in the females, in whom the clinical and laboratory manifestations are often absent, in cases with atypical presentations, and for ruling out other nephropathies and overlapping disease entities.
On light microscopy, podocytes, mesangial cells, endothelial cells, and tubular epithelial cells (predominantly distal), along with loops of Henle and collecting ducts show cytoplasmic vacuolation, which results from dissolution of accumulated glycolipid during the tissue processing. Later changes include glomerulosclerosis (segmental and/or global), interstitial fibrosis, tubular atrophy, and thickening of the vascular walls. Crystal-storing histiocytosis, histiocytic glomerulopathy in the setting of febrile illnesses, thrombotic microangiopathy, LCAT deficiency, and lipoprotein glomerulopathy, which impart a foamy appearance to the glomeruli form and yielding the differential diagnoses on light microscopy.[8]
On immunofluorescence, no immune deposits are found. In epoxy-embedded and toluidine blu-stained tissue sections, these vacuoles appear as dense dark blue cytoplasmic granules. Electron microscopy (EM) is the only pivotal method to reliably confirm or exclude FN. It shows the presence of myelin figures (or zebra bodies), which are most numerous in the podocytes, and in mesangial cells, tubular epithelial cells, especially distal, endothelial cells, and smooth muscle cells of the renal vessels. It is important to remember that these myelin figures are not pathognomic of FD and may also be seen in α-GAL inhibitors and silica-induced nephropathies, as well as other renal lipidoses (e.g., gangliosidosis GM1, Hurler's syndrome, Niemann-Pick's disease, and Farber's disease).
There are two interesting points to note in the above cases. First, these patients had variable presentations of the same disease. Thefirst case presented with lower urinary tract symptoms and milky-white urine, which acted as confounding factors. The causes of milky-white urine include chyluria (due to filariasis, malignancy, or postsurgery), proteinuria, lipiduria, hyperuricosuria, phosphaturia, and pyuria.[9] However, there was no history of prior surgery, scrotal or lower limb swelling. The patient had subnephrotic proteinuria, with the absence of phosphaturia, pyuria, or peripheral eosinophilia. The serum lipid profile was normal. The acroparesthesia in this patient was related to the damage of peripheral nerve fibers that transmit pain. On the contrary, the second case presented with a long-standing history of subnephrotic range proteinuria, and on re-examination, he was found to have skin lesions in the form of small, dark, red spots. Second, both patients had the beginning of the disease in their teens but suffered from delayed diagnosis, thus emphasizing the need for clinching the correct diagnosis.
Treatment includes ERT in the form of recombinant α-GAL enzyme or migalastat, and adjunctive therapy such as angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs). However, CKD patients require renal replacement therapy. Early initiation of the ERT at a younger age not only gives the most benefit of the therapy but also better long-term outcomes.[10] Adjunctive therapy aims at controlling proteinuria and hypertension. It may be helpful in stabilizing the kidney function or slowing of fabry's Nephropathy.[1011]
Conclusion
Possibility of a storage disorder such as Fabry disease should be kept in mind while investigating an adult with proteinuria and having foamy vacuolated appearing glomeruli on light microscopy. An ultrastructural examination is mandatory for confirmation of this rare yet characteristic histological diagnosis. It is a treatable cause of CKD, provided it is diagnosed early in the course of the disease and enzyme replacement therapy is started promptly.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that his name and initials will not be published and due efforts will be made to conceal his identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
Mrs. Madhulika Tiwari (Technical Officer, Electron Microscopy) for processing the renal biopsy tissue for ultrastructural examination.
References
- Characterization of a mutant alpha-galactosidase gene product for the late-onset cardiac form of Fabry disease. Biochem Biophys Res Commun. 1993;197:1585-9.
- [Google Scholar]
- In: The metabolic and molecular bases of inherited disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. Alpha-galactosidase A deficiency: Fabry disease. New York: McGraw Hill; 2001. p. :3733-3774.
- [Google Scholar]
- Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med 2003:138338-46.
- [Google Scholar]
- Natural history of Fabry renal disease: Influence of α-galactosidase A activity and genetic mutations on clinical course. Medicine 2002:81122-38.
- [Google Scholar]
- Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. Journal of Inherited Metabolic Disease. 2007;30(2):184-92.
- [Google Scholar]
- Renal Manifestations of Fabry disease: A narrative review? Can J Kidney Health Dis. 2021;8:2054358120985627. doi: 10.1177/
- [Google Scholar]
- Histiocytic and nonhistiocytic glomerular lesions: Foam cells and their mimickers. Am J Kidney Dis. 2016;67:329-36.
- [Google Scholar]
- Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52:353-8.
- [Google Scholar]
- Antiproteinuric therapy and Fabry nephropathy: Sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol. 2007;18:2609-17.
- [Google Scholar]




