

Translate this page into:
Glomerulocystic Kidney Disease in an Adult with DNAH1 Mutation: A Case Report

Corresponding author: Rajesh Jhorawat, Department of Nephrology, All India Institute of Medical Sciences (AIIMS), Jodhpur, India. E-mail: jhorawat2000@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Soni A, Chaturvedy M, Bajpai N, Vishwajeet V, Jhorawat R. Glomerulocystic Kidney Disease in an Adult with DNAH1 Mutation: A Case Report. Indian J Nephrol. doi: 10.25259/IJN_130_2024
Abstract
Glomerulocystic kidney disease (GCKD) is a rare cause of cystic kidney disease. It is a disease of infants and neonates, but it can also be seen in adults. We present a case of a 22-year-old married female with renal failure who was later found to have GCKD on renal biopsy.
Keywords
Glomerulocystic kidney disease
CKD
DNAH1 mutation
Primary ciliary dyskinesia
HNF-1B

Introduction
Glomerulocystic kidney disease (GCKD) is a renal cortical cystic disorder due to uniform dilation of Bowman’s space. Cysts can form from 20 micrometers to as large as 7 cm in diameter.1 Only twenty cases of GCKD have been reported in adults. We describe a case of GCKD in association with DNAH1 mutation.
Case Report
A 22-year-old woman, born of a consanguineous marriage, presented with increasing fatigue, purulent cough, and dyspnoea on exertion for one week. She had a history of nocturia, generalized fatigue, malaise, and preserved urine output with no oedematous illness Urea 94 mg/dl, Serum creatinine 4.8 mg/dl, iPTH 208 pg/ml, Vitamin D 26.7 ng/ml. She had a history of recurrent lower respiratory tract infections (LRTI) since childhood. There was no history of oliguria, dysuria, haematuria, or graveluria. Her abdomen ultrasound showed a normal pelvic-calyceal system. Three of her siblings had similar history of recurrent LRTI since childhood, and two of them had chronic kidney disease diagnosed at the age of 22 years and 19 years, respectively [Figure 1]. They both were dialysis-dependent for a year before succumbing. Physical examination was unremarkable except for severe pallor.
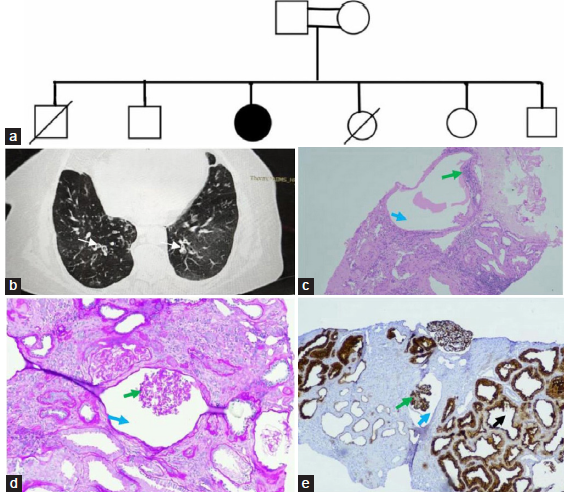
- (a) Pedigree chart (b) HRCT image showing bronchiaetatic changes (white arrows) and few ground glass opacities. Glomerular cyst at 40X (c) and 100X (d) showing rudimentary capillary tuft at one end (green arrows) of dilated Bowman’s space (blue arrows) in the subscapular area (original magnification x 400; PAS stain). (e) Immunohistochemistry with CD10 staining visceral epithelial cell and brush border epithelium of tubules Image shows a glomerular cyst with rudimentary capillary tuft at one end (green arrow) of dilated Boman's space (blue arrow) and tubular luminal dilation with simplification of the lining brush border epithelium (black arrow) (original magnification x100).
A complete urine examination revealed 2+ protein without red blood cells (RBC). On quantification, she was excreting 1.1gm of protein in her urine per day. The pulmonary evaluation revealed an obstructive pattern on the spirometer and bilateral lower lobe bronchiectasis with few ground glass opacities on the HRCT chest [Figure 1b]. To rule out primary ciliary dyskinesia, nasal biopsy, and nitric oxide tests were done but were inconclusive. Pure tone audiometry showed bilateral mild sensorineural hearing loss. Ultrasonography revealed bilateral multiple cortical cysts. Subsequently, a kidney biopsy was performed which showed prominent features of glomerulocystic change [Figure 1c-e]. Whole exome sequencing revealed a homozygous missense variant in exon 5 of the DNAH1 gene with autosomal recessive inheritance resulting in the amino acid substitution of threonine for proline at codon 240 (OMIM#617577 of Primary ciliary dyskinesia (PCD)-37) and the mutation was of unknown significance.
Discussion
Glomerulocystic kidney disease was first described in 1947 as an isolated renal abnormality, but the terminology was proposed by Taxy and Filmer in 1976.2 The pathogenesis of GCKD remains obscure. In GCKD, glomerular cysts are seen in a kidney with normal architecture without dysplasia or urinary tract obstruction.1 It can be classified into five major types, as shown in Table 1. A mutation in the HNF-1β (Hepatocyte nuclear factor-1-beta) gene has been commonly identified in the hypoplastic GCKD variant.3 It is mainly a disease of infants and newborns. Rarely has it been reported in adults presenting with the syndrome of hypertension, flank pain, and hematuria or with end-stage renal disease (ESRD) or symptomless with mild renal failure.4 An important GCKD mimicker is ARPKD, but abnormal medullary pyramids are found in the latter. Also, histology showing glomeruli with marked uniform dilatation of the Bowman’s capsule (2-3 times standard) with underlying rudimentary capillary tuft, normal tubules, and insignificant deposits on immunofluorescence suggests GCKD.2
| Familial GCKD | Autosomal dominant GCKD |
|---|---|
| Familial/sporadic heritable syndromes |
Autosomal dominant polycystic kidney disease (ADPKD) Autosomal recessive polycystic kidney disease (ARPKD) Cystic renal dysplasia Zellweger’s cerebral–renal–hepatic syndrome Tuberous sclerosis Trisomy 13 Juvenile nephronophthisis Orofacial digital syndrome type 1 Brachymeromelia renal syndrome Majewski-type short rib polydactyly syndrome Jeune’s osteodystrophy Goldston syndrome Lissencephaly |
| Syndromic, non-hereditary (GCKD as a component of other cystic diseases) |
Diffuse cystic dysplasia Renal-hepatic-pancreatic dysplasia syndrome Meckel syndrome Glutaric aciduria type 2 |
| Sporadic | New mutation |
| Acquired | Associated with HUS, obstructive uropathy |
GCKD: Glomerulocystic kidney disease, HUS: Hemolytic uremic syndrome
DNAH1 mutation has been described in male infertility and primary ciliary dyskinesia. In primary cilia, aberrant expression/signaling of HNF-1β may be indirectly related to the causation of GCKD.3 However, DNAH1 mutation is associated with infertility and GCKD; this is the first report highlighting it.5 In addition, our case is distinctive asit is more of a syndromic presentation with infertility, hearing loss, bronchiectasis, and glomerulocystic changes in the kidney, with a strong family history of kidney disease in young females. DNAH1 mutation is described in primary ciliary disorder (PCD) with infertility in males; however, in our case, it is present in females. The association of DNAH1 mutation with kidney disease through GCKD thus far is possibly a novel association of unproven significance to date. Animal models with DNAH1 mutation in ciliary disorder have been described. However, DNAH1 mutation displayed different phenotypic spectra in humans and mice.6 Hence, this mutation’s causal association in GCKD must be proven in other animal models with kidney phenotype.
Conclusion
GCKD is a rare cause of CKD, and when the syndromic presentation of PCD is there, the DNAH1 mutation may be looked at.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
References
- Familial adult glomerulocystic kidney disease. Am J Kidney Dis. 1987;9:154-65.
- [CrossRef] [PubMed] [Google Scholar]
- Glomerulocystic disease. NDT Plus. 2010;3:349-50.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Mutations in the hepatocyte nuclear factor-1 beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet. 2001;68:219-24.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Glomerulocystic disease: A case report with 10-year follow-up. Int J Pediatr Nephrol. 1982;3:321-3.
- [PubMed] [Google Scholar]
- DNAH1 gene mutations and their potential association with dysplasia of the sperm fibrous sheath and infertility in the Han Chinese population. Fertility and Sterility. 2017;107:1312-18.
- [CrossRef] [PubMed] [Google Scholar]
- Novel loss-of-function mutations in DNAH1 displayed different phenotypic spectrum in humans and mice. Front Endocrinol (Lausanne). 2021;12:1-11.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




