

Translate this page into:
Rare Case of Hemolytic Anemia and Distal Renal Tubular Acidosis in an adult due to Homozygous SLC4A1 Mutation
Address for correspondence: Dr. Wasiyeeullah Shaikh, 202, B/6, Asmita Anita Complex, Naya Nagar, Mira Road, Mumbai - 401 107, Maharashtra, India. E-mail: dr.wasishaikh@gmail.com
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
In this case study, we report an adult patient presenting with generalized weakness, marked anemia, spherocytosis, and no features of thalassemia. The patient was treated for suspicion of autoimmune hemolytic anemia but was recalcitrant to treatment. Genetic analysis revealed the patient to be homozygous for SLC4A1 c.2573C>A (p.Ala858Asp). Distal renal tubular acidosis (dRTA) can be caused by mutations in SLC4A1, which encodes the Cl−/HCO3− exchanger of the renal type A intercalated cell, kidney AE1. SLC4A1 variants have been reported in dRTA patients from North America, Europe, and Southeast Asia. In some rare instances, SLC4A1 dRTA can present with hemolytic anemia resulting in marked anemia that is not responsive to standard interventions. This report identifies an autosomal recessive inheritance pattern for SLC4A1 variants in a patient presenting with dRTA and hemolytic anemia.
Keywords
Hemolytic anemia
renal tubular acidosis

Introduction
Individuals with hereditary distal renal tubular acidosis (dRTA) typically present as infants, although later presentation is possible, especially in individuals with mild or hypomorphic SLC4A1 variants. Initial clinical manifestations can also include polyuria, polydipsia, gastrointestinal disorder, decreased appetite, and dehydration. In these patients, evaluation of blood biochemistry reveals hyperchloremic non–anion gap metabolic acidosis and hypokalemia. Renal complications of dRTA include nephrocalcinosis, nephrolithiasis, and impaired renal function. Additional manifestations include bone demineralization (rickets, osteomalacia) and rarely hereditary hemolytic anemia.
Case Report
We report a case of a 29-year-old married female, born of a nonconsanguineous marriage, residing in Mumbai, India, who presented with generalized weakness, fatigue, malaise, and dyspnea on exertion. On examination, she had cushingoid features, vitals were stable, and pallor was present but no evidence of jaundice. Abdominal examination revealed moderate splenomegaly, but the rest of the systemic examination was normal. On laboratory evaluation, she had anemia with hemoglobin (Hb) of 7 g/dL, and a peripheral blood smear revealed spherocytes. The patient was evaluated in detail for anemia wherein she had an elevated reticulocyte count of 3.02%. Her serum haptoglobin levels, bilirubin, and lactate dehydrogenase levels were normal, whereas the direct Coombs test (DCT) and the indirect Coombs test were negative. Also, her Hb electrophoresis and G6PD (glucose-6-phosphate dehydrogenase) were normal, and vitamin B12 was within the range. Immunological workup for ANA (antinuclear antibody), dsDNA (double-stranded deoxyribonucleic acid), and APLA (antiphospholipid antibody) were negative, and heavy metal screening for lead and copper was within the reference range. The laboratory values are listed in Table 1.
| Test | Value | Test | Value |
|---|---|---|---|
| Hb | 7 g/dL | Ca2+ | 7.9 mg/dL |
| WBC | 3,790 cells/mm3 | PO4 | 2.7 mg/dL |
| Platelets | 152,000 cells/mm3 | Uric acid | 4.7 mg/dL |
| Reticulocyte count | 3.02% | PTH | 137 pg/mL |
| Haptoglobin | 10 mg/dL | Alkaline phosphatase | 103 IU/L |
| LDH | 397 IU/L | Copper | 163 µg/dl |
| Total bilirubin | 1.35 mg/dL | Lead | 2.07 µg/dl |
| Indirect bilirubin | 0.77 mg/dL | TSH | 1.4 mIU/l |
| Creatinine | 1.43 mg/dL | ESR | 102 mm/h |
| Na+ | 137 mEq/L | Vitamin B12 | 680 pg/ml |
| K+ | 2.97 mEq/L | ||
| Cl− | 108 mEq/L | ||
| HCO3− | 12 mEq/L |
Hb=Hemoglobin; WBC=White blood cell; PTH=Parathyroid hormone; TSH=Thyroid stimulating hormone; ESR=Erythrocyte sedimentation rate; Ca=Calcium; PO4=Phosphate; Na=Sodium; K=Potassium; Cl=Chlorine; HCO3=Bicarbonate
Bone marrow aspiration showed normocellular marrow with the presence of ring sideroblasts (80%). The iron store was not evaluated due to lack of particles. Bone marrow biopsy showed predominantly superficial bony trabeculae, and the marrow spaces were normocellular to mildly hypercellular. FISH (fluorescence in situ hybridization) for myelodysplastic syndrome (MDS) was normal. X-ray of the right hip joint had evidence of sclerosis in the right acetabulum with a reduction in joint space. Ultrasound of the abdomen revealed moderate splenomegaly with multiple shadows in both kidneys suggestive of nephrocalcinosis and small-sized right kidney.
The patient was treated with blood transfusions, folic acid, and pyridoxine (vitamin B6) supplementation, which stabilized the patient’s hemoglobin at 9.5 g/dL. The patient was started on intravenous steroids and oral immunosuppression with azathioprine and mycophenolate mofetil due to suspicion of autoimmune hemolytic anemia. It was noted on the subsequent follow-up that the hemoglobin level had dropped to 5.2 g/dL despite oral steroids and immunosuppression, requiring blood transfusions. In view of persistent hemolytic anemia and spherocytosis refractory to the standard line of treatment with immunosuppression, the patient was advised to undergo splenectomy after appropriate immunization. Nephrology opinion was sort in view of acidosis on metabolic workup and presence of bilateral nephrocalcinosis with right small-sized kidney on sonography.
On inquiry, she gave a history of limping of the right leg due to bone pain and repeated renal calculi requiring multiple urological interventions. Apart from metabolic acidosis, the patient had low serum calcium and elevated parathyroid hormone. Urine was alkaline with a pH of 8, whereas the serum pH was 7.2. The patient’s parents and siblings had no clinical history of hematological or renal disease, and hematological indices of parents were unremarkable. Given the findings of normal anion gap metabolic acidosis with alkaline urinary pH, diagnosis of renal tubular acidosis was made. Treatment with oral sodium bicarbonate and potassium citrate was commenced, which resolved the acidosis, and the patient did well post-splenectomy and oral bicarbonate supplementation.
The presence of combined hemolytic anemia and dRTA suggested the evaluation of erythroid band 3/AE1. Focused exome sequencing demonstrated the presence of an apparently homozygous pathogenic variant in SLC4A1 (NM_000342.4:c.2573C>A (p.Ala858Asp)), which was previously reported in a patient with a similar phenotype.[1]
Discussion
Our patient presented with recurrent symptomatic anemia requiring blood transfusions. Prior to referral, spherocytosis was seen on smear, but DCT was negative. The patient exhibited a rapid drop in hemoglobin each time posttransfusion. The patient also had splenomegaly, and hence a diagnosis of hereditary spherocytosis (HS) was considered. However, both her parents had normal hematological parameters and smear findings. Hence, it was considered that she had DCT negative AIHA (autoimmune hemolytic anemia) and was initiated on steroids, to which she responded well with subsequent reduction in the need for blood transfusion. A bone marrow examination was performed in view of autoimmune hemolysis to rule out hematological malignancy. The aspirate was normocellular with 80% ringed sideroblasts. Bone marrow biopsy showed bony trabeculae, and all markers for autoimmunity were negative.
In view of steroid dependence that led to cushingoid features and behavioral changes, it was decided to perform splenectomy, to which she had an excellent response in terms of restoring normal hemoglobin without the requirement of blood transfusions. For the sideroblasts, she was given large doses of vitamin B6. With the clinical picture of nephrocalcinosis, normal anion gap acidosis with alkaline urine a diagnosis of renal tubular acidosis was made. In asymptomatic patients with no family history, this condition may go unnoticed. But genetic testing determined that distal renal tubular acidosis and spherocytosis were caused by a missense mutation in the SCL4A1 gene.
dRTA is a clinical syndrome characterized by impairment of acid secretion by type A intercalated cells in the distal nephron and collecting tubule, resulting in decreased net acid excretion and the inability of the kidney to lower urinary pH <5.5 in spite of spontaneous academia or after acid loading.[2-4] The decreased net acid excretion attributable to decreased ammonium and titratable acid excretion leads to a positive acid balance and hyperchloremic metabolic acidosis. The presence of chronic metabolic acidosis causes hypocitraturia and hypercalciuria creating a favorable environment for urinary stone formation and nephrocalcinosis. The other associated abnormalities include hypokalemia leading to muscle weakness and metabolic bone disease.[4]
The defects in the transport pathways involving the acid–base transporters of type A intercalated cells in medullary collecting duct leading to an increase in apical membrane permeability that causes dRTA include H+ ATPase, H+ K+ ATPase, HCO3−–Cl− exchanger, H+ back leak, and carbonic anhydrase type II as shown in Figure 1.[5] Most studies suggest that inherited forms of dRTA are due to defects in basolateral HCO3−–Cl− exchanger (SLC4A1) or subunits of H+ ATPase (ATP6V1B1 or ATP6V0A4). Inherited forms of dRTA can have autosomal dominant or autosomal recessive inheritance patterns.[6]
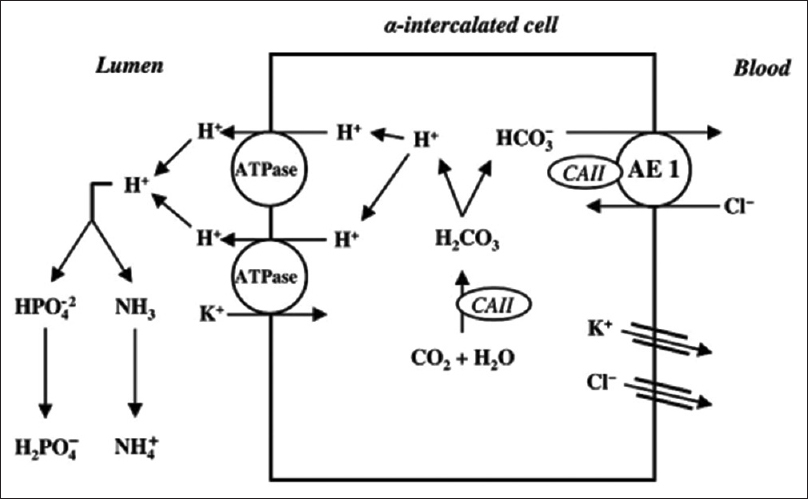
- Schematic model of the α-intercalated cell and the H+ secretion in cortical collecting tubule
Autosomal dominant dRTA occurs due to heterozygous mutations affecting SLC4A1, which encodes Cl−/HCO3− exchanger (AE1). This is the predominant inheritance pattern for SLC4A1 dRTA. It is predominantly expressed in the erythrocytes (eAE1) and in the kidney (kAE1). kAE1 mediates an electroneutral exchange of chloride for bicarbonate at the basolateral membrane of type A intercalated cells.[7,8] eAE1 is involved in the facilitation of red cell metabolism and maintenance of erythrocyte structural stability.[8] The majority of AE1 mutations apparently cause only erythroid abnormalities without renal phenotype, and hence most cause autosomal dominant forms of HS and are not encountered in homozygous form, suggesting embryonic lethality.[9] Based on our findings and those of others,[5] autosomal recessive inheritance of dRTA is possible in presumably mild SLC4A1 variants like c.2573C>A.
Autosomal recessive dRTA occurs due to mutations in the proton pump in type A intercalated cells. ATP6V1B1 gene encodes the B1 subunit of H-ATPase expressed apically intercalated cells and in the cochlea and endolymphatic sac leading to dRTA with deafness[10,11] [Table 2]. Since there was no history and findings in the patient’s parents, this was a case of a missense mutation of SCL4A1 mutation leading to hemolytic anemia with dRTA. Our patient was homozygous for SLC4A1 mutation, and to the best of our knowledge, there are only two case reports in Indian patients with combined hemolytic anemia and dRTA who shared homozygous mutations of the SLC4A1 gene, and both these patients had presented in childhood.[12] There is another case report of seven pediatric Omani Arab subjects, with dRTA and hemolytic anemia and homozygous SLC4A1 mutation, all of whom are of the pediatric age group.[1]
| Dominant RTA | Recessive RTA | Osteopetrosis with RTA | |
|---|---|---|---|
| Gene | SLC4A1 | ATP6V1B1 | CA2: Carbonic anhydrase II |
| ATP6V0A4 | |||
| Presentation | Adult | Infancy | Childhood |
| Nephrolithiasis | Nephrocalcinosis | Nephrocalcinosis | |
| Nephrocalcinosis | Vomiting/dehydration | Thickened bones | |
| Growth impairment | Mental retardation | ||
| Hearing loss | |||
| Hematological | Secondary erythrocytosis | - | Anemia |
| Biochemistry | Hyperchloremic acidosis | Hyperchloremic acidosis | Hyperchloremic acidosis |
| Hypokalemia | Hypokalemia | Hypokalemia | |
| Urine | pH >5.5 | pH >5.5 | pH <5.5 |
| Hypercalciuria | Hypercalciuria |
dRTA=Distal renal tubular acidosis; RTA=Renal tubular acidosis
Conclusion
This report indicates that SLC4A1 dRTA with hemolytic anemia can present with delayed-onset phenotype in patients, despite commonly presenting in infancy. Reporting this patient provides additional evidence for the variable phenotypes in some cases that are important for medical providers to understand when evaluating adult patients with symptoms of dRTA. Furthermore, this report indicates a successful medical intervention for patients presenting with such a phenotype. Early treatment as indicated here will help reduce the morbidity of dRTA, as was the case with our patient.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- dRTA and hemolytic anemia: First detailed description of SLC4A1 A858D mutation in homozygous state. Eur J Haematol. 2012;88:350-5.
- [Google Scholar]
- Hereditary distal renal tubular acidosis: New understandings. Annu Rev Med. 2001;52:471-84.
- [Google Scholar]
- Trafficking defect of mutant kidney anion exchanger 1 (kAE1) proteins associated with distal renal tubular acidosis and Southeast Asian ovalocytosis. Biochem Biophys Res Commun. 2006;350:723-30.
- [Google Scholar]
- Defects in processing and trafficking of the AE1 Cl-/HCO3- exchanger associated with inherited distal renal tubular acidosis. Clin Exp Nephrol. 2004;8:1-11.
- [Google Scholar]
- Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71-5.
- [Google Scholar]
- Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84-90.
- [Google Scholar]
- Hemolytic anemia and distal renal tubular acidosis in two Indian patients homozygous for SLC4A1/AE1 mutation A858D. Am J Hematol. 2010;85:824-8.
- [Google Scholar]




