

Translate this page into:
IGg4-related Disease Presenting as Rapidly Progressive Renal Failure and Inguinal Iymphadenopathy
Address for correspondence: Dr. Maniyar Iqbal Anvar, Department of Nephrology, VIMS, Cantonment, Bellary, Karnataka - 583 104, India. E-mail: anvarmd@yahoo.co.in
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
A 58-year-old man presented with swelling in the left inguinal region and loss of appetite, nausea and azotemia with a serum creatinine of 5.2 mg/dL and urine albumin of 1+ and normal size kidneys with no hematuria. His serum creatinine increased to 9 mg/dL over 1 month, his total proteins were 10.8 and serum albumin was 3.3 g/dL, and lymph node excision biopsy showed fragments of lymphoid tissue with interfollicular areas containing sheets of plasma cells and atretic germinal centers. In view of unexplained renal failure, he underwent renal biopsy, which showed interstitial inflammatory infiltrate composed of lymphocytes and rich in plasma cells with storiform fibrosis and a possibility of IgG4-related renal disease was reported. On further evaluation, serum electrophoresis showed no M band. Serum IgG4 levels were 18.2 g/L (0.03–2 g/L). A diagnosis of IgG4-related renal disease was confirmed.
Keywords
IgG4
lymphadenopathy renal failure
plasma cells

Introduction
The IgG4-related disease is an increasingly recognized immune-mediated condition that comprises a collection of disorders that share pathologic, serologic and clinical features.[1] The commonly shared features are the involvement of different organs like the pancreas, lymph nodes, salivary glands, lacrimal glands, retroperitoneal fibrosis, renal involvement, orbital involvement. The pathologic features involving all organs will include tumor-like swelling of involved organs, lymphoplasmacytic infiltrate with IgG4-positive plasma cells and varying degrees of fibrosis called “storiform” fibrosis [Figure 1]. The renal involvement is usually in the form of tubulointerstitial nephritis (TIN),[2] sometimes membranous nephropathy and crescentic glomerulonephritis will occur.

- Typical morphology of Ig G4 Related tubulointerstitial nephritis. Interstitial compartment shows storiform pattern of fibrosis (Trichrome stain, original magnification x10) whereas glomeruli appear unaffected except for the periglomerular fibrosis.
Case Report
We report a case of 58 years old male who presented with pain and swelling of left inguinal region and history of nausea, loss of appetite, epigastric pain, belching. On examination, we found left inguinal lymphadenopathy, and his blood pressure of 130/80 mmHg and a pulse of 70 per minute. Upon investigation, he was found to have Hb of 9.1 g/dL, Tc of 5400 cells/cumm, platelet count 1.76 lakhs, urea 100 mg/dL, serum creatinine 5.2 mg/dL, which increased to 9 mg/dL over a month's period, the other investigations were sodium 138 meq/L, potassium 4.1 meq/L, chloride 102.6 mmol/L, serum total proteins 10.8 g/dL, serum albumin 3.9 g/dL. Urine examination showed albumin 1+, 1–2 pus cells/HPF, 1-2 EPI Cells/HPF, 0–1 RBCs/HPF and spot protein/creatine ratio of 1.3 and Bence-Jones protein negative. His serum electrophoresis showed no M band. His kidneys were sized RK 8.9*4.3 cm and LK was 9.2*4.8 cm, and there were multiple conglomerated hypoechoic enlarged lymph nodes in left iliac fossa and mild prostate enlargement. The patient underwent surgical excision biopsy of Left iliac lymph node and after few days he went ultrasound-guided renal biopsy. The lymph node biopsy showed fragments of lymphoid follicles, few atretic germinal centers containing hyaline material and interfollicular areas containing sheets of plasma cells along with focal vascular proliferation. Occasional eosinophils were noted. A diagnosis of Castleman's disease - plasma cell variant was reported. Subsequently, the renal biopsy sample contained total 14 glomeruli of which, three were globally sclerosed, six showed perihilar segmental sclerosis with periglomerular fibrosis but no mesangial or endocapillary hypercellularity, crescent, or necrosis. The basement membrane showed focal thickening and wrinkling but no spikes. The tubules and interstitium showed severe chronic damage and storiform fibrosis [Figure 1] with a mixed inflammatory infiltrate consisting of abundant infiltrate of plasma cells [Figure 2] along with lymphocytes and few polymorphs. The immunofluorescence microscopy of the renal biopsy was negative for IgG, IgA, IgM, C3, C1q, Kappa, and lambda within the glomeruli. A possibility of IgG4-related renal disease was reported with advice for further evaluation with serum IgG4 levels and Immunohistochemistry for IgG4+/IgG+ plasma cells. Further investigations showed high serum IgG4 levels of 18.2 g/L (Ref Range: 0.03–2.0), 9 times the normal value. A diagnosis of IgG4-related disease was confirmed. Reviving the literature we got complements levels of the patient measured which showed both C3 (34.1 mg/dL) and C4 (2.9 mg/L) levels below normal which further confirmed our diagnosis as hypocomplementemia occurs with renal involvement in IgG4 related disease. To complete the workup, immunohistochemistry for IgG and IgG4 was performed and the ratio IgG4/IgG was found to be 86% (>40%, according to the diagnostic criteria by the Japanese IgG4 team) [Figure 3]. We started the patient on prednisolone at 0.6 mg/kg/day in a single daily dose, after which his serum creatine decreased from 11 mg/dL to 8.1 mg over a week and continued to decrease further. On further follow-up, the patient's serum creatinine decreased to 2.9 mg/dL after 6 months and the patient is currently on follow-up at monthly intervals with 20 mg prednisolone per day. Apart from inguinal lymphadenopathy and renal involvement and high proteins with normal albumin and hypocomplementemia there was no involvement of salivary glands, orbital tissue or pancreas.
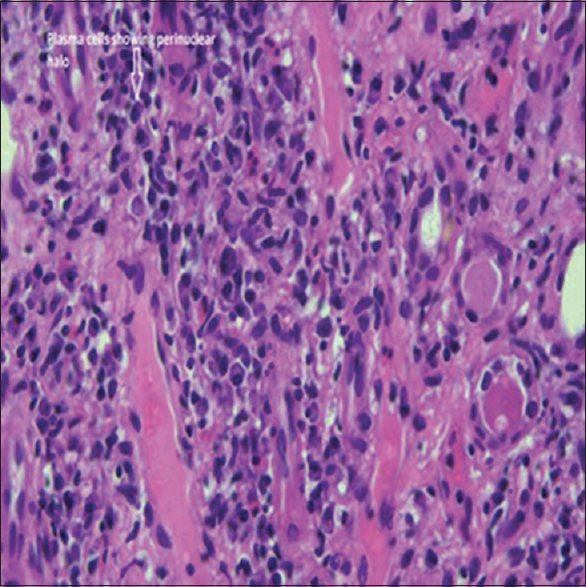
- Typical pathological feature encountered depicts mononuclear interstitial infiltrate rich in plasma cells along with focal areas of tubulitis (Haematoxylin and Eosin stain, original magnification x40)
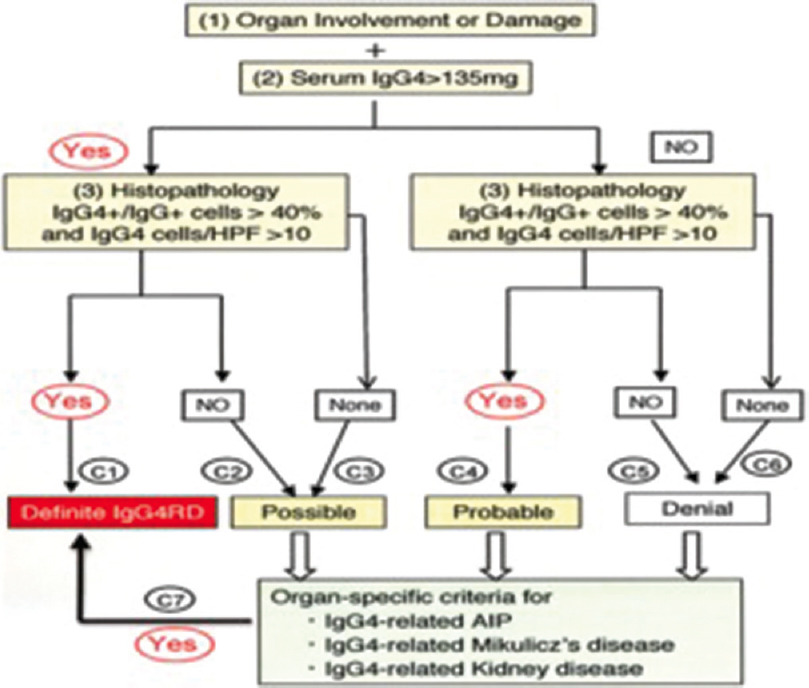
- Comprehensive diagnostic criteria for IGg4-related disease
The 2 diagnostic criteria for IgG4-related disease in our patient were
-
Japanese society of Sjogren's syndrome 2008 IgG4 related Mickuliz disease, for a subset of IgG4-related kidney disease, were histological features of dense lymphoplasmacytic infiltration by >10 IgG4+ plasma cells/HPF and IgG4/IgG+ plasma cells of >40% and characteristic features surrounding the nest of lymphocytes/plasma cells and serum IgG level >135 mg/dl, hypocomplementemia and other findings of lymphadenopathy.
-
Comprehensive diagnostic criteria of IgG4-RD which consists of concept-diagnostic criteria and explanatory notes and validated in studies of IgG4 MD, IgG4RD, and IgG4 AIP were
-
Organ involvement and damage
-
Serum IgG+ >135 mg/dL
-
As both were positive and histopathology of renal biopsy had IgG4/IgG Plasma cells >40% (our case had 86%), which satisfied the criteria for organ-specific criteria of IgG4-related kidney disease.
Discussion
Mikulicz's disease, the former term for IgG4-related renal disease, was first reported by Dr. Johann von Mikulicz in 1892, defining it as enlargement and mononuclear infiltration of salivary glands (lacrimal, parotid and submandibular). Subsequently, it was associated with Sjogren syndrome and was described as the elevation of serum IgG4 concentration and tissue infiltration by IgG4-positive cells. Thereafter, various nomenclatures such as IgG4-related autoimmune disease, IgG4-associated multifocal systemic fibrosis, IgG4-related systemic disease, systemic IgG4 plasmacytic syndrome (SIPS), and IgG4-related multiorgan lymphoproliferative syndrome (Ig G4-MOLPS).
IgG4- related disease was first recognized as a cause of autoimmune pancreatitis in 2001 and was reported as a cause of TIN in 2004 (first reported). The IgG4-related disease primarily affects older men,[3] which was similar in our patient who was male aged 58 years.
The major presentation of this disorder which affects more than one organ, include:
-
Type 1 (IgG4-related) autoimmune pancreatitis (AIP)
-
Salivary gland enlargement or sclerosing sialadenitis. A combination of the lacrimal, parotid, and submandibular enlargement formerly (Mikulicz syndrome). isolated submandibular swelling Kuttner tumor.
-
Orbital disease, causing proptosis due to lacrimal gland enlargement, extraocular muscle involvement, or orbital pseudotumor
-
Retro peritoneal fibrosis
-
Renal involvement in the form of TIN.
Reviewing the literature, apart from the histologic feature of infiltration of involved tissue with IgG4 positive plasma cells, the serum levels of IgG 4 levels are elevated (defined as >135 mg/dL, >86 mg/dL, or >121 mg/dL depending on laboratory) in two-thirds of patients.[2]
A good initial therapeutic response to glucocorticoids is obtained if extensive tissue fibrosis has not supervened.[4] IgG4 related disease (IgG4RD) has a slight predominance for middle-aged and older males. In our case, the patient was male 57 years. This male predilection is more important for conditions of AIP, IgG4-related TIN, and retroperitoneal fibrosis as in our case which had TIN. But in Head and Neck involvement like IgG4-related ophthalmic disease and IgG4-related sialadenitis males and females appear equally affected.[3] Although the disease occurs more commonly in men, the disease extent and severity appear similar in men and women. In a series of 125 patients with biopsy-proven IgG4-RD, the number of organs involved, the degree of serum IgG4 elevation, and damage from IgG4-RD did not differ in a male and female patient.[5] The pathogenesis of IgG4-RD is poorly understood, findings consistent with both allergic and autoimmune disorders are present.[26] Elevations in serum and tissue IgG4 Concentrations are not specific to IgG4-RD, they are also found in disorders like multicentric Castleman disease, eosinophilic granulomatosis with polyangiitis (Churg-Strauss), sarcoidosis and a large number of other conditions.[4]
The CD4+ cytotoxic T cells are the most abundant cells in affected tissues. These clonally expanded populations of cells are central to the pathogenesis of the disease.[7] They produce IL1, TGF β, and interferon-gamma which mediate fibrosis. A T follicular Helper response different from CD4+ cytotoxic T cells will form germinal centers within lymph nodes and involved organs and produce IL-4 which causes IgG4 class switch, culminating in IgG4 secreting plasmablasts and long-lived plasma cells. Even though IgG4 is a multisystem disease we would describe lymph node and renal involvement.
Lymphadenopathy is usually observed with other manifestations of disease or maybe only clinical manifestation.[8] Since biopsies of lymph nodes are difficult to interpret it is advisable to do another organ biopsy as we did in our case by doing Renal biopsy. The lymphadenopathy is non-tender, rubbery, and multiple group involvement is there like mediastinal, hilar, intraabdominal, axillary node, but in our case, we had inguinal involvement which was a little rare. In a study of 114 patients with various organ involvement, lymphadenopathy was present in 41% of patients.[9]
Five histologic patterns may be seen which features abundant IgG4-positive cells: The majority have eosinophil infiltration. The patterns are
Type I.- Multicentric Castleman disease like
Type II- Follicular hyperplasia
Type III- Interfollicular expansion
Type IV- Progressive transformation of a germinal center-like
Type V – Nodal inflammatory pseudotumor like
Our patient had type I Multicentric Castleman's disease like histology.
Renal disease
In individual case and case series described in IgG4-RD the most common finding is TIN.[10] Patients are middle age or elderly as in our case, the histologic changes are lymphoplasmacytic infiltration of renal interstitium and fibrosis. Immunohistochemistry showing increased IgG4 positive Plasma cells. In a retrospective, a multicentric Japanese study of 153 patients with suspected of IgG4 RD, 23 patients (15%) had TIN due to IgG4-RD, and among 23 patients (96% of TIN) had multiorgan involvement.[11] The extrarenal manifestation are sialadenitis (83%), lymphadenopathy (44%) AIPC (39%), dacryoadenitis (30%) lung lesion (26%). Other than TIN 3 of 23 patients had mesangioproliferative glomerulonephritis, one membranous and focal segmental endocapillary hypercellularity.
Patients with IgG4 Related TIN are profoundly hypocomplementemia, as our patient had low C3 (34. mg/dL) and C4 (2.9 mg/dL). IgG4 binds poorly to complement, hence, other IgG subclass IgG1 and IgG3 are responsible for its activation. In another retrospective study from Australia, IgG4-related TIN represented 1% of total biopsies exhibiting TIN which was independent of glomerular disease.[12] IgG4 related membranous nephropathy is much less common than TIN, but these two complications sometimes occur together.[14] In a series of nine patients with IgG4-related membranous GN, five patients had concurrent IgG4-RD.[13] None of the patients with IgG4-RD were positive for phospholipase A2 receptor on biopsy.
Diagnostic criteria
Since one specific criterion could not be used comprehensive criteria consisting of three segments: concept diagnostic criteria, explanatory notes, and validated in studies of IgG4MD, IgG4-RD, and IgG4-AIP. An algorithm developed as a combination of comprehensive diagnostic criteria and organ-specific criteria is shown below [Figure 3].
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published, and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- IgG4-related sclerosing disease, a clinical appraisal of an evolving clinicopatholologic entity. Adv Anat Pathol. 2010;17:303-32.
- [Google Scholar]
- A clinical overview of IgG4 related systemic disease. Curr Opin Rheumatol. 2011;23:57-66.
- [Google Scholar]
- IgG4-related disease: Clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67:2466-75.
- [Google Scholar]
- Two critical genes (HLA-DRB1 and ABCF1) in the HLA region are associated with the susceptibility to autoimmune pancreatitis. Immunogenetics. 2007;59:45-52.
- [Google Scholar]
- Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol. 2016;138:825-38.
- [Google Scholar]
- Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:671-81.
- [Google Scholar]
- IgG4-related disease: A cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34:1812-9.
- [Google Scholar]
- Autoimmune pancreatitis associated with renal lesions mimicking metastatic tumours. CMAJ. 2006;175:367-9.
- [Google Scholar]
- Mercury-induced membranous nephropathy: Clinical and pathological features. Clin J Am Soc Nephrol. 2010;5:439-44.
- [Google Scholar]
- The incidence of IgG4-positive plasma cells staining TIN in patients with biopsy-proven tubulointerstitial nephritis. J Clin Pathol. 2017;70:483-7.
- [Google Scholar]
- Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21-30.
- [Google Scholar]




