

Translate this page into:
Late-Onset Bartter’s Syndrome Type II with End-Stage Renal Disease Due to a Novel Mutation in KCNJ1 Gene in an Indian Adult Male – A Case Report
Address for correspondence: Dr. Sree Bhushan Raju D, Department of Nephrology, First Floor, Near ARCU, Nizam’s Institute of Medical Sciences, Punjagutta, Hyderabad - 500 082, Telangana, India. E-mail: sreebhushan@hotmail.com
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Mutations in ROMK1 potassium channel gene (KCNJ1) causes antenatal/neonatal Bartter’s syndrome type II, which presents with renal salt wasting, hypokalemic metabolic alkalosis, secondary hyperaldosteronism, hypercalciuria, and nephrocalcinosis. We herein describe a case of late-onset Bartter’s syndrome type II with progressive renal failure requiring renal replacement therapy secondary to a novel homozygous missense mutation in Exon 2 of KCNJ1 gene (c.500G>A). With this case, we aim to highlight the need for a high index of suspicion and the role of genetic evaluation to diagnose clinically unclassified cases of nephrocalcinosis with renal electrolyte abnormalities more so in late and atypical presentations.
Keywords
Bartter’s syndrome
KCNJ1
late-onset
ROMK

Introduction
Bartter’s syndrome is an autosomal recessive disorder affecting the function of the thick ascending limb of the loop of Henle giving a clinical picture of salt wasting and hypokalemic metabolic alkalosis. Most cases present antenatally or in neonates, and it has an estimated prevalence of 1 pmp.[1] Bartter’s syndrome is classified as type 1–type 6, which reflects the gene affected. Type 1 is caused by mutations in SLC12A1, type 2 by a mutation in KCNJ1, type 3 by CLC-Kb mutation, type 4 by BSND mutation, type 5 by activating mutation in CaSR, and type 6 by mutations in both CLC- Ka and CLC-Kb.[2] [Figure 1] We report a case of late-onset Bartter’s syndrome type II due to a novel mutation in KCNJ1 gene with progressive renal failure.

- Schematic representation of transport proteins involved in Bartter’s syndrome. An epithelial cell of the thick ascending limb of the Loop of Henle with Na+–K+–2Cl− cotransporter and renal outer medullary potassium channel (ROMK) at the apical side. Chloride channel ClC-Ka and ClC-Kb with its Barttin subunit, calcium-sensing receptor (CaSR), and Na+–K+ ATPase channel at the basolateral side
Case Details
A 34-year-old gentleman came with chief complains of nausea, vomiting, and decreased appetite for 10 days. On probing further, he revealed that he had history of polydipsia, polyuria, nocturia, and repeated episodes of carpopedal spasms in his childhood. He was born via normal vaginal delivery at full term with an uneventful antenatal or neonatal period. The patient denied history of paralysis/paresis of limbs, recurrent urinary tract infections, fractures, hearing disturbances, and visual loss. There was no history of renal diseases in the family. He was found to have a creatinine of 2 mg% 4 years back and was under follow-up of a nephrologist at a local hospital.
On physical examination, the patient was moderately built and well-nourished with short stature (height: 140 cm) and no other obvious deformities or skeletal dysmorphic facies. He was normotensive (120/80 mm Hg) with normal ECG and 2D Echo. Laboratory tests revealed severe azotemia (urea: 139 mg%, creatinine: 10 mg%). There was hypokalemia (3.28 meq/L), hypocalcemia (5.0 mg%), and hyperphosphatemia (7.0 mg%) with normal sodium (140 meq/L) and chloride (96 meq/L) levels. Vitamin D levels were low (20 ng/ml) with inappropriately normal PTH levels (90 pg/ml). In arterial blood gas analysis, pH was normal and bicarbonate was low with respiratory compensation (pH = 7.41, serum bicarbonate = 18 mmol/l). The evaluation of 24-hour urinary examination was hampered in view of decreased residual renal function with a urine output of approximately 100 ml/day. His immunological profile (ANA, dsDNA, Anti Sm, and Anti Ro/La) and viral markers (HIV, HbsAg, and HCV) were negative. X-ray of the lumbosacral spine and hands were unremarkable other than for mild changes of renal osteodystrophy.
Ultrasound abdomen was suggestive of medullary nephrocalcinosis with normal-sized kidneys, which was confirmed by computed tomography scan. [Figure 2]. A percutaneous kidney biopsy was performed from the lower pole of the left kidney, which revealed 1/6 glomeruli showing segmental sclerosis, a large area of interstitial fibrosis, and diffuse dense infiltration of lymphocytes and plasma cells in the interstitium. Tubules showing intraluminal calcification [Figure 3] along with the destruction of tubules and unremarkable vessels.

- CT scan of the abdomen showing medullary nephrocalcinosis
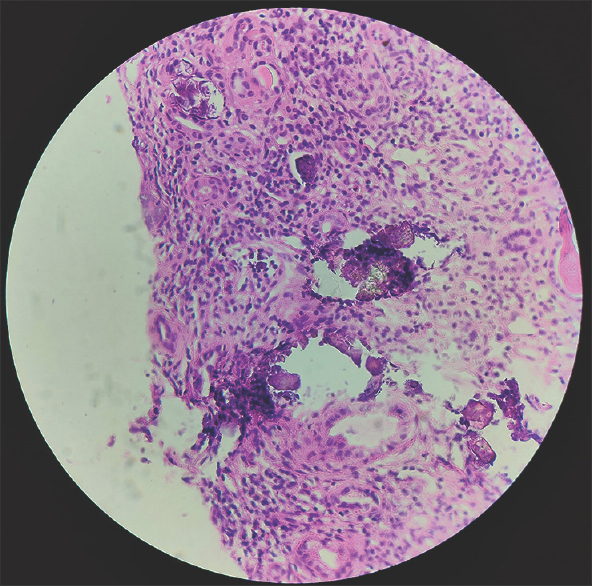
- Interstitial inflammation comprising of lymphocytes and plasma cells with multiple foci of intratubular calcification (Hematoxylin and Eosin stain, magnification: ×100)
The patient was initiated on hemodialysis via a temporary right internal jugular venous catheter, and transplant evaluation was initiated. After informed consent, genomic DNA was extracted from peripheral blood leukocytes and clinical exome sequencing was performed. The analysis revealed a homozygous missense variation in exon 2 of KCNJ1 gene in the chromosome 11 that results in the amino-acid substitution of glutamic acid for glycine at codon 167. {c.500 G>A, homozygous, exon 2, codon 167, p.Gly167Glu}.
Discussion
Inactivating loss of function mutations in the KCNJ1 gene encoding the apical potassium inwardly rectifying outer medullary potassium channel (ROMK) in the thick ascending limb of Henle is classically associated with antenatal/neonatal Bartter’s syndrome type II. The ROMK (Kir 1.1) channel generates the lumen positive potential, which is critical for transepithelial Na+- Cl- transport via the NKCC2 channel in the thick ascending limb.[3]
More than 40 cases of KCNJ1 mutations have been described so far. Most of the mutations are missense/nonsense mutations substituting conserved amino acid residues, predominantly within coding exon 2, the most influential putative functional domain of ROMK.[4] In our case, a novel homozygous missense mutation in the codon 167 of exon 2 of the KCNJ1 gene was found, which is known to result in defective potassium conductance due to a conformational change in the ROMK channel,[5] which impairs the function of NKCC2 and leads to salt wasting tubulopathy.
However, the KCNJ1 mutation in the present case could represent a mild phenotype considering the lack of classical antenatal/neonatal onset of the disease. Table 1 lists the various novel mutations seen in similar case reports of late-onset Bartter’s syndrome. The present case had a short stature, probably indicating the tubular loss of salts and calcium since adolescence. The growth retardation in Bartter’s syndrome has been attributed to many causes such as hypokalemia, depletion of extracellular fluid volume, malnutrition, polydipsia, polyuria, salt wasting, hypercalciuria, and growth hormone deficiency.[9] Progressive renal failure in Bartter syndrome is extraordinarily rare. Lin et al.[10] described a case of chronic renal failure in a 17-year-old Chinese boy with classical Bartter’s syndrome due to a novel mutation in CLC-Kb coding for the chloride channel.
| Author | Age/Sex | eGFR (ml/min) | Mutation |
|---|---|---|---|
| Gollasch et al.[6] | 43/F | 58 | Compound heterozygous missense mutation/in Exon 2 of KCNJ1(c. 197T>A and c.875G>A |
| Hwang L, et al.[7] | 35/M | 45 | Homozygous missense mutation in KCNJ1/(c. 658C>T). |
| Khandelwal P, et al.[8] | 13/F | 110 | Compound heterozygous missense mutation in/Exon 2 of KCNJ1(c. 146G>A and c.657C>G) |
| Present Case | 34/M | 6 | Homozygous missense mutation in Exon 2 of/KCNJ1(c.500 G>A). |
Possible mechanisms of renal failure in Bartter’s syndrome are 1) chronic hypokalemic nephropathy, which is associated with impaired renal angiogenesis;[11] 2) volume depletion activating the renin–angiotensin aldosterone system (RAAS); 3) renin–angiotensin–II, aldosterone system contributing to renal fibrosis by triggering the proliferation of transforming growth factor-beta (TGF-b) and plasminogen activator inhibitor-1 gene expression and accelerate renal damage by induction of reactive oxygen species.[12] The presence of chronic interstitial fibrosis in the renal biopsy of the patient probably suggests an interplay of the various abovementioned mechanisms leading to progressive renal failure and the intratubular calcification seen in the biopsy may also have contributed to progressive renal damage[13] in the present case.
Based on the above mentioned mechanisms, Walsh et al.[14] suggested that long-term blockade of mineralocorticoid receptor in BS with spironolactone or eplerenone (or perhaps even direct renin inhibition or angiotensin-converting enzyme inhibitors) may be justified more for renal protection in Bartter’s patients than for trying to correct hypokalemia per se.
To the best of our knowledge, this is the first case of late-onset Bartter’s syndrome type II with progressive renal failure secondary to a novel KCNJ1 mutation being reported in the Indian literature.
Conclusion
With this case, we aimed to highlight the need for a high index of suspicion and the role of genetic evaluation in diagnosing clinically unclassified cases of nephrocalcinosis with renal electrolyte abnormalities, more so in atypical and late presentations. The evaluation of different pathogenic mechanisms of ROMK mutation will also help in tailoring distinct rescue mechanisms according to the underlying genetic defect.[15]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592-9.
- [Google Scholar]
- Reabsorption of sodium chloride--Lessons from the chloride channels. N-Engl J Med. 2004;350:1281-3.
- [Google Scholar]
- Presence of luminal K+, a prerequisite for active NaCl transport in the cortical thick ascending limb of Henle's loop of rabbit kidney. Pflugers Arch. 1981;392:92-4.
- [Google Scholar]
- Expanding the spectrum of genetic mutations in antenatal bartter syndrome type-II. Pediatr Int. 2013;55:371-73.
- [Google Scholar]
- Mutations in the ROMK gene in antenatal Bartter syndrome are associated with impaired K+channel function. Biochem Biophys Res Common. 1997;230:641-5.
- [Google Scholar]
- Nephrocalcinosis as adult presentation of Bartter syndrome type-II. Neth J Med. 2014;72(2):91-3.
- [Google Scholar]
- Isolated nephrocalcinosis due to compound heterozygous mutations in renal outer medullary potassium channel. CEN Case Rep. 2020;9:232-6.
- [Google Scholar]
- Growth hormone deficiency in children with antenatal bartter syndrome. J-Pediatr Endocrinol Metab. 2019;32:225-31.
- [Google Scholar]
- Chronic renal failure in a boy with classic Bartter's syndrome due to a novel mutation in CLCNKB coding for the chloride channel. Eur J Pediatr. 2009;168:1129-33.
- [Google Scholar]
- Hypokalemic nephropathy is associated with impaired angiogenesis. J-Am Soc Nephrol. 2008;19:125-34.
- [Google Scholar]
- Local angiotension II and transforming growth factor beta-1 in renal fibrosis in rats. Hypertension. 2000;35:1078-84.
- [Google Scholar]
- Nephrocalcinosis:Molecular insights into calcium precipitation within the kidney. Clin Sci-(Lond). 2004;106:546-61.
- [Google Scholar]
- Does hypokalemia cause nephropathy?An observational study of renal function in patients with Bartter or Gitelmann syndrome. QJM. 2011;104:939-44.
- [Google Scholar]
- Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. 2003;64:923-32.
- [Google Scholar]




