

Translate this page into:
A Novel Mutation in GATA3 Gene in a Case of Hypoparathyroidism, Deafness, and Renal Dysplasia Syndrome
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
A 39-year-old male was incidentally detected to have hypertension and chronic kidney disease (CKD) with left solitary functioning kidney in 2017. He has bilateral sensorineural hearing loss since adolescence. He was initially suspected to have adynamic bone disease in view of low parathyroid hormone levels and was started on teriparatide injections and calcium supplements. Despite all these measures, he had persistent hypocalcemia and low parathyroid hormone levels. Hence, Hypoparathyroidism, Deafness, and Renal dysplasia (HDR) syndrome was suspected, and the patient was evaluated for the same. Genetic analysis revealed the presence of a de novo and a novel frameshift mutation in GATA-binding protein 3 (GATA3) gene on chromosome 10p. To the best of our knowledge, this is the first case report of HDR syndrome being diagnosed by genetic analysis in India.
Keywords
Adynamic Bone Disease
deafness and kidney
GATA3 gene
HDR syndrome

Introduction
Hypoparathyroidism, Deafness, and Renal dysplasia (HDR) syndrome (Barakat syndrome) is characterized by the triad of hypoparathyroidism, sensorineural deafness, and renal dysplasia.[12] It was named HDR syndrome by Hasegawa et al.[3] (OMIM# 146255). It is a rare autosomal dominant genetic disorder caused by haploinsufficiency of GATA-binding protein 3 (GATA3) gene on chromosome 10p14.[4] We describe a case of HDR syndrome, proven by genetic analysis, in a young adult who was previously suspected to have adynamic bone disease (ABD). He has a de novo and a novel mutation in the GATA3 gene, which has not been previously described in the literature.
Case Report
A 39-year-old male presented to us in February 2017 with incidentally detected hypertension and renal dysfunction (serum creatinine of 2.7 mg/dl). There was no history of pedal edema, dysuria, oliguria, or hematuria. Also, there was no history of fever, joint pain, or rashes. No history of nonsteroidal anti-inflammatory drug (NSAID) abuse, any alternate medication intake, or addictions was reported. He has bilateral hearing impairment since adolescence. On examination, his blood pressure was 150/90 mmHg. Systemic examination was normal except bilateral hearing loss. There was no family history of kidney disease.
Investigations done in the initial visit confirmed renal dysfunction (serum creatinine 3 mg/dl) with normal electrolytes, hypocalcemia, and hyperphosphatemia (serum calcium 5.4 mg/dl, serum phosphorous 6.3 mg/dl, serum albumin 4.1 g/dl, serum alkaline phosphatase [ALP] 89 IU/ml). Urine albumin was 2+, and there were no active urinary sediments. Ultrasound abdomen and pelvis showed the presence of only left kidney measuring 9.9 cm in length, cortical thickness of 1.3 cm, with increased cortical echogenicity. Right kidney was not visible. Audiometry confirmed the presence of moderate degree of bilateral sensory neural hearing loss. A provisional diagnosis of chronic kidney disease (CKD)- Stage IV, left solitary functioning kidney, with CKD-mineral bone disease was made. He was started on tablet calcium carbonate 1.5 g/day, tablet calcitriol 0.25 μg/day, and tablet sevelamer 400 mg thrice per day on out patients basis and was advised to review with serum parathyroid hormone (PTH) levels.
Four months later (July 2017), he was found to have an improvement in biochemistry (serum calcium 9.5 mg/dl, serum phosphorous 5 mg/dl, ALP 83 IU/ml), but a very low PTH (serum PTH 9.2 pg/ml [normal range 15–65 pg/ml]). He was suspected to have ABD; calcitriol and phosphate binders were stopped and he was started on injection teriparatide (rPTH 1-34) 20 μg subcutaneous once a day. He was compliant with his medications. His serum creatinine remained stable throughout, but there was persistent hypocalcemia and hypoparathyroidism. Computed tomography (CT) brain did not show any basal ganglia calcification. The bone scan was normal. His calcium requirement was almost 4 g/day, and he was continued on teriparatide injection 20 μg/day. The investigations done annually have been tabulated in Table 1.
| Feb 2017 | July 2017 | 2018 | 2019 | 2021 | |
|---|---|---|---|---|---|
| Hemoglobin (g/dl) | 13.2 | 14.4 | 12.8 | 13.6 | 13.9 |
| Total counts (cells/dl) | 8550 | 8270 | 6700 | 13700 | 7130 |
| Platelet counts (cells/dl) | 3.3 lakhs | 2.73 lakhs | 2.8 lakhs | 2.63 lakhs | 3.19 lakhs |
| Blood urea nitrogen (mg/dl) | 50 | 27 | 24 | 25 | 23 |
| Serum creatinine (mg/dl) | 2.7 | 3.2 | 3.2 | 3.08 | 3.2 |
| Serum calcium (mg/dl) | 5.3 | 9.5 | 6.4 | 7.0 | 6.5 |
| Serum phosphorous (mg/dl) | 6.2 | 5.0 | 6.0 | 4.8 | 4.8 |
| Serum albumin (g/dl) | 4.1 | 4.0 | 4.0 | ||
| Serum alkaline phosphatase (IU/ml) | 89 | 83 | 72 | 67 | 70 |
| PTH (pg/ml) | 9.2 | 9.0 | 10.6 | 13.6 | |
| Serum vitamin D (IU/ml) | 52 | 44 | 43.5 | 39 | |
| Serum TSH (m IU/l) | 3.76 | 3.28 | |||
| Serum uric acid (mg/dl) | 5.9 | 6.2 | |||
| Urine albumin (by dipstick) | 2+ | 2+ | 2+ | ||
| Plan | Calcium, calcitriol, and sevelamer added | Impression: ? adynamic bone disease Calcium, calcitriol, and sevelamer stopped | Calcium and teriparatide added | Calcium, teriparatide continued CT brain was normal DEXA scan was normal | Patient had stopped teriparatide for 3 months Advised to restart |
CT=computed tomography, DEXA=dual-energy X-ray absorptiometry, PTH=parathyroid hormone, TSH=thyroid stimulating hormone
In view of persistent hypocalcemia and hypoparathyroidism, in the background of CKD- Stage IV, left solitary functioning kidney, and bilateral hearing loss, a relook at our diagnosis of ABD was done. A possibility of HDR syndrome was considered. Patient underwent genetic counselling and was tested specifically for GATA3 gene mutation. Next-generation sequencing was done, and he was found to have heterozygous frameshift variant, c681delC; P. Glu228SerfsTer38, in exon 3 of GATA3 gene. Sanger sequencing also confirmed the same [Figure 1]. It was diagnosed as pathogenic as per the American College of Medical Genetics and Genomics (ACMG) guidelines. This is a novel mutation and has not been described earlier. Thus, our patient was diagnosed to have HDR syndrome/Barakat syndrome.
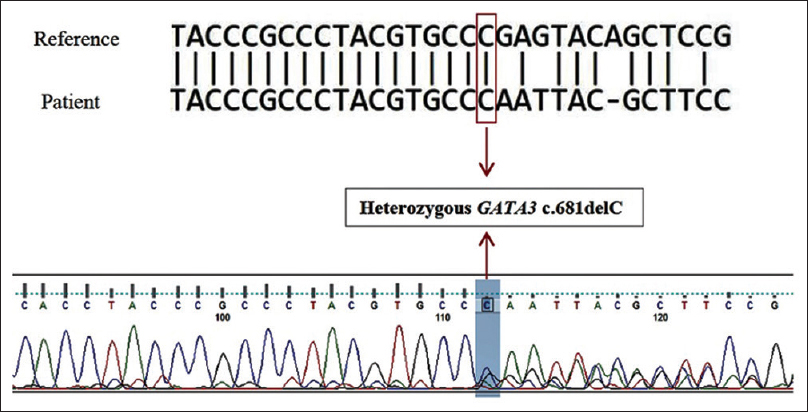
- The blue highlighted area in the electropherogram shows a deletion of a nucleotide “C” in one allele (heterozygous deletion). Thus the electropherogram and NCBI nucleotide BLAST sequence shows disrupted from the position of the heterozygous nucleotide deletion
On retaking the family history, he was found to be the youngest among the four siblings, born out of a nonconsanguineous marriage. No history of hearing impairment among the family members was reported. He is married and has three children, with none of them have hearing impairment. None of the family members have undergone any formal testing for kidney ailments. Thus, our patient has a de novo mutation in the GATA3 gene. In view of CKD- Stage IV, he has been advised to continue teriparatide injection and calcium supplements. His family members have been counselled about the need for further evaluation, blood tests, and genetic testing to diagnose the disease at the earliest.
Discussion
HDR syndrome/Barakat syndrome is characterized by the triad of hypoparathyroidism, sensorineural deafness, and renal dysplasia.[12] This was first described in 1977 by Barakat et al.[1] in two twin brothers with steroid-resistant nephrotic syndrome, bilateral sensory neural hearing loss, and hypoparathyroidism. It is a rare autosomal dominant genetic disorder caused by haploinsufficiency of GATA3 gene on chromosome 10p14.[4] GATA3 protein is one of the six members of the GATA family of transcription factors which are involved in the embryonic development of parathyroid glands, kidney, inner ear, thymus, and central nervous system (CNS).
A systematic review of 155 case reports of HDR syndrome published in various journals found hypoparathyroidism and deafness in 95% of patients and renal abnormality in only 60% of patients.[5] Deafness is the characteristic manifestation, which is bilateral, symmetrical sensorineural deafness. The degree of hearing loss and the age of onset may vary. Hypoparathyroidism has variable age of onset. Patients can have asymptomatic or symptomatic hypocalcemia, with undetectable or low serum PTH levels. Renal anomalies can vary. Patients can have renal aplasia, dysplasia, hypoplasia, and vesicouretric reflux. Some case reports of diffuse proliferative glomerulonephritis and focal segmental glomerulosclerosis have also been described.[6]
Our patient had moderate bilateral sensorineural hearing loss, which started in his adolescence. He complained of occasional fatigue and tiredness, but never had any classical complains of hypocalcaemia like muscle twitching or seizures. He was incidentally detected to have hypertension and CKD in the fourth decade of life. Based on these, HDR syndrome was suspected in our case. There are a few case reports of clinically diagnosed HDR syndrome diagnosed among Indian patients.[78] In a case report published in Indian Journal of Nephrology by Singh et al.,[9] the patient was clinically diagnosed to have HDR syndrome associated with hyperkalemia and renal tubular acidosis. Genetic analyses have not been done in these cases. To the best of our knowledge, our patient is the first case of HDR syndrome being diagnosed in India by genetic analysis.
There are 51 mutations of GATA3 gene that have been described in literature.[10] Our patient had a heterozygous frameshift variant, c. 681delc; pGlu228SerfsTer38, in exon 3 of GATA3 gene. The variant is not reported in population database (gnomAD, 1000 genomes) and is a novel mutation. It is predicted to be damaging based on the in silico analysis (Genomic Evolutionary Rate Profiling [GERP] score and Mutation Taster). It has been classified as pathogenic as per the ACMG guidelines. This has also been submitted to the ClinVar database, but we are yet to receive ClinVar submission ID. A summary of phenotypic and genotypic characteristics of the present case and previously reported cases of HDR syndrome has been presented in Table 2.
| Our case | Singh et al.[9] | Meena et al.[8] | Anne et al.[11] | Chen et al.[12] | Gul et al.[13] | Okawa et al.[14] | |
|---|---|---|---|---|---|---|---|
| Gender | Male | Female | Male | Male | Male | Male | Female |
| Age | 39 years | 39 years | 20 years | 13 years | 19 years | 13 years | 33 years |
| Clinical features | No symptoms | Seizures | Seizures | Tetany | Tetany | Tetany | Seizures |
| Serum calcium levels (mg/dl) | 5.4 | 3.9 | 3.4 | 6.4 | 7.4 | 6.7 | 5.2 |
| Serum phosphorous level (mg/dl) | 6.3 | 10.5 | 7 | 8 | 5.3 | 9 | 4.86 |
| Intact PTH (pg/ml) | 9.2 | 0.23 | 8.61 | 7.1 | 5 | 20 | 7 |
| Sensory neural deafnessa | Moderate | Moderate | Severe | Moderate | Moderately severe | Moderately severe | Mild |
| Renal anomaly | Solitary left kidney | Solitary right kidney | Solitary right kidney | Nil | Small kidneys | Small left pelvic kidney | Right renal dysplasia |
| GATA3 gene mutation | Exon 3 C681delC; P Glu228 SerfsTer38 | Not done | Not done | Whole gene deletion of GATA3 | Exon 2 c. 529dupCp. Arg177profs X126 | Exon 4 p.R276Q c. 827G>A | Exon 4 p.R299Q |
PTH=parathyroid hormone. aDegree of hearing loss: normal <25 dB, mild 26–40 dB, moderate 41-55 dB, moderately severe 56-70 dB, and profound >90 dB
ABD is more prevalent among dialysis patients, especially among diabetics and those who are on CAPD, but it is not uncommon to see it in CKD- stages 3–5, with a prevalence rate of about 18%.[15] ABD is a state of relative hypoparathyroidism. Bone-specific ALP is the most useful marker of bone formation, and if elevated, it helps us to exclude ABD. There is no evidence about the diagnostic value of any biochemical marker of bone remodelling (e.g., N-telopeptide, osteocalcin, etc.) as a predictor of bone histomorphometry among CKD or dialysis patients.
Our patient was initially suspected to have ABD as he had very low PTH levels. Bone histomorphometry evaluation was not done. Despite stopping calcitriol, phosphate binders, and initiating teriparatide injections, there was no improvement in the calcium and PTH levels. In the background of CKD-solitary functioning left kidney, early-onset sensorineural hearing loss, and persistently low PTH levels despite all therapies, we reconsidered our diagnosis of ABD. Genetic analysis has helped us in confirming our clinical diagnosis of HDR syndrome.
Our patient has been advised to continue calcium supplements and teriparatide injections in view of CKD- Stage IV- solitary functioning kidney. Calcitriol has not been started as it may lead to hypercalciuria and nephrocalcinosis. All his siblings and children have been advised to undergo evaluation to diagnose this disease at the earliest.
Conclusion
As far as we know, this is the first case report of HDR syndrome in India proven by genetic analysis. Our patient has a novel and a de novo mutation in the GATA3 gene. There should be a high index of clinical suspicion of a genetic ailment, especially if the patient has both kidney and ear involvement. Genetic analysis will help us in diagnosing the disease entity at the earliest and enable us to counsel the patient regarding the prognosis of the disease in the long term.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Familial nephrosis, nerve deafness, and hypoparathyroidism. J Pediatr. 1977;91:61-4.
- [Google Scholar]
- Brief report: Autosomal dominant familial hypoparathyroidism, sensorineural deafness and renal dysplasia. N Eng J Med. 1992;327:1069-74.
- [Google Scholar]
- HDR syndrome (hypoparathyroidism, sensorineural deafness, renal dysplasia) associated with del (10)(p13) Am J Med Genet. 1997;73:416-8.
- [Google Scholar]
- Clinical and mutational spectrum of hypoparathyroidism, deafness and renal dysplasia syndrome. Nephrol Dial Transplant. 2017;32:830-7.
- [Google Scholar]
- Renal phenotypic variability in HDR syndrome: Glomerular nephropathy as a novel finding. Eur J Pediatr. 2013;172:107-10.
- [Google Scholar]
- Hypoparathyroidism, deafness and renal failure: A case of Barakat syndrome. Eur J Mol Clin Med. 2020;7:5723-25.
- [Google Scholar]
- Hyperkalemia unveiled: A case of Barakat syndrome. Indian J Nephrol. 2020;30:135-6.
- [Google Scholar]
- The first Korean case of HDR syndrome confirmed by clinical and molecular investigation. Yonsei Med J. 2015;56:300-3.
- [Google Scholar]
- Hypoparathyroidism Sensorineural deafness and renal disease (Barakat syndrome) caused by a reduced gene dosage in GATA3: A case report and review of literature. BMC Endocr Disord. 2019;19:111.
- [Google Scholar]
- Identification of a novel de novo GATA3 mutation in a patient with HDR syndrome. J Int Med Res. 2015;43:718-24.
- [Google Scholar]
- Novel de novo GATA binding protein 3 mutation in a Turkish boy with Hypoparathyroidism, deafness and renal dysplasia syndrome. J Clin Res Pediatric Endocrinol. 2015;7:344-8.
- [Google Scholar]
- A novel loos of function mutation in GATA3 (p. R299Q) in a Japanese family with Hypoparathyroidism, deafness and renal dysplasia syndrome. BMC Endocr Disord. 2015;15:66-8.
- [Google Scholar]
- KDIGO clinical practice guideline for diagnosis, evaluation, prevention, and treatment of chronic kidney disease- mineral bone disorder (CKD-MBD) Kidney Int Suppl. 2009S1;113
- [Google Scholar]




