

Translate this page into:
Light chain deposition disease in a postrenal transplant patient
Address for correspondence: Dr. Kusum Joshi, Department of Histopathology, Post-graduate Institute of Medical Education and Research, Chandigarh - 160 012, India. E-mail: kus_joshi@yahoo.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The morphological spectrum of light chain deposition disease (LCDD) may range from normal glomerular morphology to mesangio-proliferative to mesangio-capillary to nodular sclerosing patterns. Due to the inconsistencies regarding treatment and the universally poor graft outcome of post-transplant LCDD, it is imperative to maintain a high index of suspicion and perform relevant investigations for clinching this diagnosis. A 40-year-old lady was diagnosed as a case of membrano-proliferative glomerulonephritis 3 years back, for which she underwent a live unrelated renal allograft transplant. Postoperative period was complicated by an acute rise in serum creatinine on the 21st postoperative day. Biopsy showed patchy acute cortical necrosis, which responded to conservative management. The present admission was for renal failure and subnephrotic proteinuria. A kidney biopsy was performed, and all the 14 glomeruli examined showed a mesangiocapillary pattern of glomerular injury with cellular nodule formation in some. The nodules were PAS and Congo red negative. Immunofluorescence showed glomerular and tubular basement staining for Kappa light chains only. Electron microscopy showed the characteristic granular deposits in subendothelial location in the glomerulus, and in tubular basement membranes, thus confirming the diagnosis of LCDD. Membranoproliferative pattern of glomerular injury in the pre- and posttransplant setting has a wide range of differential diagnoses; LCDD being one of them.
Keywords
Light chain deposition disease
postrenal transplant kidney
glomerulus

Introduction
Pure light chain deposition disease (LCDD) in kidney is classically described by the presence of nodular glomerulosclerosis by light microscopy, monoclonal deposits of light chains within renal basement membranes by immunofluorescence, and electron-dense, granular deposits along basement membranes by electron microscopy.[1] Clinical manifestations may range from nephrotic syndrome (in 30–50% of patients), <1 g/day proteinuria (~25% patients), progressive renal insufficiency (in approximately 70% of patients), and hypertension (in more than 80% of the patients) among others.[2] Morphologically, the accumulation of immunoglobulin light chains in the kidney may involve the glomeruli, tubules, or the interstitium and vessels, exclusively or concurrently, and generate a wide spectrum of pathologic findings that may resemble many diseases, including minimal change disease, diabetic glomerulosclerosis, amyloidosis, membranoproliferative glomerulonephritis type 1, and nonspecific interstitial fibrosis and tubular atrophy.[13–5] The importance of differentiating these conditions from LCDD cannot be overemphasized. The therapy is different, so is the prognosis and patient survival. Despite the limited data available, LCDD invariably recurs in the graft with poor graft survival. Thus, the more or less universal agreement on discouraging renal transplant in LCDD makes it mandatory for it to be diagnosed in a pretransplant patient with renal failure. The present case exemplifies this fact.
Case Report
Herein we present a case of a 43-year-old lady, non-diabetic, non-hypertensive, who underwent a live unrelated renal allograft 3 years back. Renal biopsy done elsewhere was reported as chronic glomerulonephritis possibly “Mesangiocapillary glomerulonephritis” (glass slides of the pretransplant biopsy could not be retrieved). There was no evidence of any other organ system dysfunction at the time of transplant. On day 21 after transplant, she reported to our hospital for an acute increase in serum creatinine to 3.2 mg/dl from a base line of 1.9 mg/dl. The urine output was reduced with mild proteinuria (<1 g/day). An allograft biopsy was performed which showed infarction of a part of the cortical tissue. Stain for acid fast bacilli and fungi (Grocott's stain) did not show any organisms. The noninfarcted area consisted of eight glomeruli which were unremarkable, so were the tubules, interstitium, and blood vessels. No thrombosed vessels were seen. The glomeruli or blood vessels did not show any changes of thrombotic microangiopathy. There was no evidence of rejection or drug overdose. C4d stain was performed and was negative in the peritubular capillaries. Cyclosporine levels were within normal limits. Renal function improved over a period of 10 days, and at the time of discharge, her serum creatinine was 1.2 mg/dl, urine output was adequate, and there was no proteinuria. She was continued on triple immunosuppression (Tacrolimus, MMF, and prednisolone) and was followed up in the outpatient department for a year which was largely uneventful, following which she was lost to follow-up. Twenty-six months after the transplant, she presented with worsening renal allograft function and diarrhoea. The diarrhea responded to broad-spectrum antibiotics and thus was bacterial in origin. However, the renal function deteriorated rapidly. Her serum creatinine increased from 3.2 to 5.6 mg/dl in a period of 1 week with a proteinuria of 1.5 g/day. Urine microscopy showed 5–10 RBCs per high-power field; no casts or crystals were seen. A renal biopsy was done which had a single linear core with 14 glomeruli. Majority of the glomeruli showed lobular expansion by cellular nodules of varying sizes accompanied by thickening of the glomerular capillary wall. The nodules were composed of endocapillary proliferation and mesangial hypercellularity which was global in some, and focal in rest. Three glomeruli showed only uniform glomerular capillary wall thickening devoid of any proliferation. Overall, the findings were consistent with a “mesangiocapillary pattern of glomerular injury.” The nodules were pale on periodic acid Schiff stain and were Congo red negative. Masson's trichrome stain failed to show any fuschinophilic immune complex deposits in the glomerular mesangium and/or capillary walls. Also, the tubular basement membranes (TBMs) of nonatrophic tubules appeared thicker and refractile even in the areas devoid of any tubulointerstitial scarring, and this finding was confirmed on Masson's trichrome stain. Immunohistochemistry was performed for Kappa and Lamba antibodies which showed preferential staining of the TBMs and glomerular capillary walls by Kappa antibodies only. Tissue was submitted only for light microscopy, thus immunofluorescence was done from the paraffin-embedded tissue using the proteinase K digestion method, and EM using the standard protocols for formalin-fixed tissue. Immunofluorescence showed marked staining of the glomerular capillary walls along with TBMs by Kappa antibody. In contrast. Lambda was positive in a few casts only. There was no deposition of any other antisera (IgG, IgA, IgM. and C3) studied. Electron microscopy showed the characteristic dark, granular deposits in the subendothelial location in the glomerulus and focally in the tubular basement membranes, thus confirming the diagnosis of LCDD [Figure 1]. Subsequent investigations revealed a monoclonal “M” spike in both the urine and serum electrophoresis. Serum-free light chain assay was elevated with a markedly elevated Kappa: Lambda ratio. Bone marrow examination showed 15% plasma cells; however, no lytic lesions were seen on the bone scans. On follow-up, patient's renal function deteriorated further, she became oliguric, and had to be started on maintenance hemodialysis.
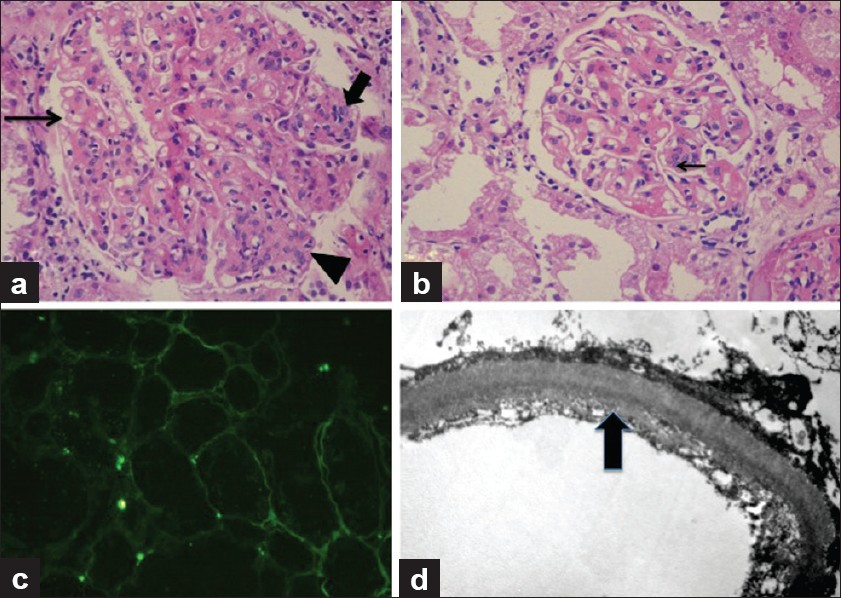
- A panel of photomicrographs of light microscopy (a and b, H and E, ×100), immunofluorescence (c, H and E, ×50), and electron microscopy (d, H and E, ×5000) of the renal biopsy of the index case. (a) A glomerulus showing mesangiocapillary pattern with mesangial hypercellularity and nodule formation (arrow head), focal endocapillary proliferation (bold arrow), and glomerular capillary wall thickening with focal splitting (fine arrow). (b) Another glomerulus showing predominantly capillary wall thickening with only an occasional focus of mesangial hypercellularity (arrow) and no nodularity. (c) Immunoflorescence for Kappa antisera showed marked positivity in the tubular basement membranes. Similar positivity was also seen in the glomerular capillary walls. Lambda antisera was negative in these locations and so were other immunoglobulin antisera (IgG, IgM, and IgA). (d) Ultrastructure photomicrograph showing granular electron-dense deposits along the subendothelial region of the glomerular capillary wall which are diagnostic of MIDD
Discussion
LCDD was first described about three decades ago,[6] and since then, the reported incidence of LCDD is quite low. However, due to varied clinical presentations and many differential diagnoses on morphology; it is possibly both underrecognized and underreported. Advanced renal failure requiring dialysis is a common finding at presentation.[7] The most common pattern on morphology is nodular mesangial sclerosis, which is preceded by other morphological manifestations, including mesangial and membranoproliferative patterns, and even minimal change disease.[45] A high index of suspicion and pretransplant screening of patients for LCDD by serum and urine electrophoresis is the only way to clinch the diagnosis. As the upper limit of age for kidney transplantation has increased, increasing number of patients with monoclonal gammopathy of undertermined significance may be encountered. The importance of pretransplant diagnosis lies in the experience gained in posttransplant patients with LCDD. Only a few patients with LCDD, both diagnosed and undiagnosed pretransplant, have undergone renal transplant.[7–9] The notion of freedom from the disease and increasing graft survival was behind the idea of kidney transplant in diagnosed cases of LCDD. Although limited to a few studies, the results obtained are universally discouraging. The disease recurs in the allograft in at least 80% of patients with a mean recurrence interval of 5–50 months after transplant.[78] The graft-to-recurrence interval in the index case was 26 months. In one of the largest single-center experience of renal transplantation in patients with LCDD, of seven posttransplant patients, four died after the transplant due to graft complications, two after going on dialysis, and the remaining three died due to complications of the disease with graft still functioning.[9] The median reported survival rates after transplant are 18 months to 5 years, that too, with considerable morbidity.[9] Short, et al.[8] reviewed six of their own cases of LCDD and one case from the case report by David-Neto.[9] The latter case, which is an exception rather than the rule, was the only one with a 44 months recurrence-free period. Several factors predic poorer outcome, including age, initial serum creatinine, serum calcium, types of immunoglobulin deposits, and types of lesions seen on renal biopsy.[1] Overall, given the downhill course of LCDD in posttransplant patients, renal transplant is to be discouraged by all means. Although it cannot be confirmed that the index case is a recurrence of LCDD in the graft kidney rather than a de novo occurrence, however, considering the limited availability of immunofluorescence, and unavailability of electron microscopy in most of the private sector establishments in our country, the proposal is justified. So take-home messages from this case are that the morphological spectrum of LCDD has a wide range of differential diagnosis, resulting in potential under/misdiagnosis. It invariably recurs in the posttransplant period with a high potential of graft failure/death of the patient, hence kidney transplant is to be avoided unless measures (pre- and posttransplant) can be taken to control the production of abnormal light chains.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Renal monoclonal immunoglobulin deposition disease: The disease spectrum. J Am Soc Nephrol. 2001;12:1482-92.
- [Google Scholar]
- Kidney and liver involvement in monoclonal light chain disorders. Semin Nephrol. 2002;22:319-30.
- [Google Scholar]
- Organized deposits in the kidney and look-alikes. Ultrastruct Pathol. 2003;27:295-312.
- [Google Scholar]
- Light chain deposition disease of the kidney: Morphological aspects in 24 patients. Virchows Arch. 1994;425:271-80.
- [Google Scholar]
- Spectrum of glomerular and tubulointerstitial renal lesions associated with monotypical immunoglobulin light chain deposition. Lab Invest. 1991;64:527-37.
- [Google Scholar]
- Long-term outcome of renal transplantation in light-chain deposition disease. Am J Kidney Dis. 2004;43:147-53.
- [Google Scholar]
- Recurrence of light chain nephropathy in a renal allograft.A case report and review of the literature. Am J Nephrol. 2001;21:237-40.
- [Google Scholar]
- Renal transplantation in systemic light-chain deposition (SLCD): A 44 month follow-up without recurrence. Transplant Proc. 1989;21:2128-9.
- [Google Scholar]




