

Translate this page into:
Mitochondrial Neurogastrointestinal Encephalomyopathy: A Nonrenal Indication for Peritoneal Dialysis
Address for correspondence: Dr. Ram, Department of Nephrology, SVIMS, Tirupati, Andhra Pradesh, India. E-mail: ram_5_1999@yahoo.com
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Mitochondrial neurogastrointestinal encephalomyopathy is a rare autosomal recessive disorder characterized by severe muscle wasting, gastrointestinal dysmotility, leukoencephalopathy, peripheral neuropathy, and ophthalmoplegia. The pathogenesis involves the accumulation of very high concentrations of nucleosides dThd and dUrd along with depletion of nucleotide dCTP. One of the treatment measures is the removal of nucleosides dThd and dUrd by hemodialysis and peritoneal dialysis. Only a few patient reports of dialysis as a measure to remove nucleosides had been reported.
Keywords
dCTP
dThd
dUrd
hemodialysis
mitochondrial neurogastrointestinal encephalomyopathy
peritoneal dialysis
thymidine phosphorylase deficiency

Introduction
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a rare autosomal recessive disorder characterized by severe muscle wasting, gastrointestinal dysmotility, leukoencephalopathy, peripheral neuropathy, and ophthalmoplegia.[1] The mutations in the TYMP gene encoding thymidine phosphorylase (TP) cause alterations in the respiratory chain and mitochondrial DNA (mtDNA).[2] The prognosis of this disease is limited; most patients die by the age of 35 years, but survival may range from 15 to 54 years.[1]
Case Report
A 20-year-old male patient had normal birth history and milestones until 2 years of age. Delayed milestones appeared from the 3rd year along with failure to thrive. There was no mental retardation or cognitive impairment at any time. There were no seizures, gait abnormalities, bowel or bladder disturbances, or visual and hearing difficulties. At 10 years of his age, he had acute abdomen for which appendicectomy was done. However, the abdominal pain recurred frequently. The abdominal pain in the beginning was one or two episodes a week; later before, he attained the age of 16 years it worsened to more than 5–10 episodes a day. He also developed early satiety and postprandial vomiting. There was no difficulty in swallowing. He had weight loss and progressive generalized wasting of muscle [Figures 1 and 2]. He had numbness of extremities. It worsened when he was stationary. He did not develop any secondary sexual characters nor there any growth spurt. He had a normal younger sibling. There was no similar family history.
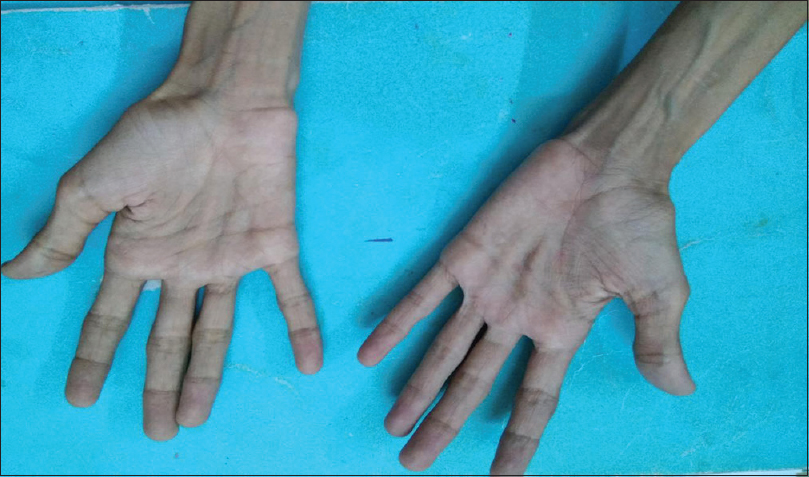
- Wasting of palmar eminences
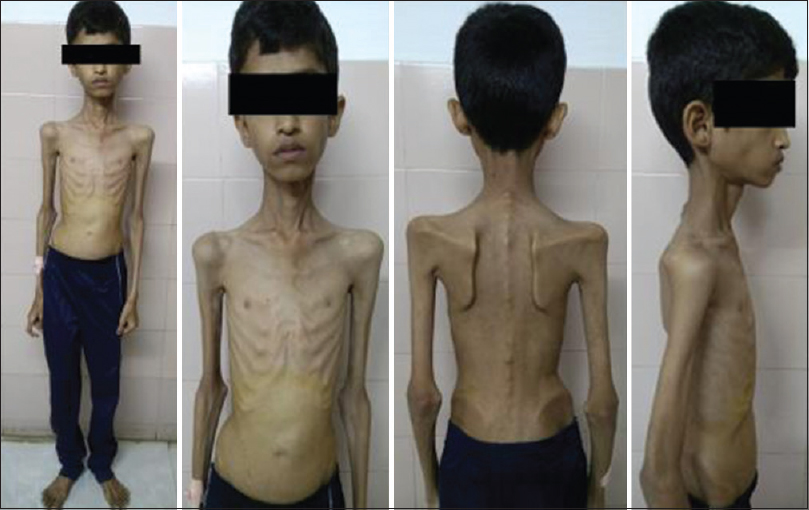
- Generalized muscle wasting
On examination, the height of the patient was 147 cm, weighed 18 kg with a body mass index of just 8.33 kg/m2 and body surface area of 0.92 m2. He had severe generalized muscle wasting. His gait was normal. Lumbar lordosis was apparent when he was walking. His pulse was 84/min, noninvasive blood pressure of 100/60 mmHg. There were no thyromegaly, organomegaly, generalized lymphadenopathy, and bone tenderness. Neurological examination showed normal mental functions. Cranial nerve examination was suggested by chronic progressive external ophthalmoplegia with normal fundus.
Power in upper and lower limbs was 4/5 MRC grading. Deep tendon reflexes were sluggish (1+). Sensory system examination was showing loss of joint proprioception with other modalities being normal. There were no cerebellar or long tract signs. Skull examination was normal in size and shape. Spine examination demonstrated lumbar scoliosis to the left. Cardiovascular and respiratory system was unremarkable. External genitalia showed both descended testes in the scrotum with no pubic hair. Penis was small with normal external meatus and normal glans. Axillary hair, beard, and moustache were absent.
He was admitted to neurology department after he had one episode of convulsion. On magnetic resonance imaging (MRI) brain, T2 signal hyperintensities were seen involving in both frontal, temporal, parietal, and occipital lobes along with pons and both cerebellar hemispheres. [Figure 3] As these features were indicated a mitochondrial leukoencephalopathy, the possibility of MNGIE was suspected. He was further evaluated by gene sequencing for TYMP gene, the culprit gene for MNGIE. On gene sequencing, a frameshift mutation along with deletion of exon 4 was noted, which is novel mutation in TYMP gene and hence establishing the diagnosis of MNGIE. Serum levels of thymidine or deoxyuridine were not done.
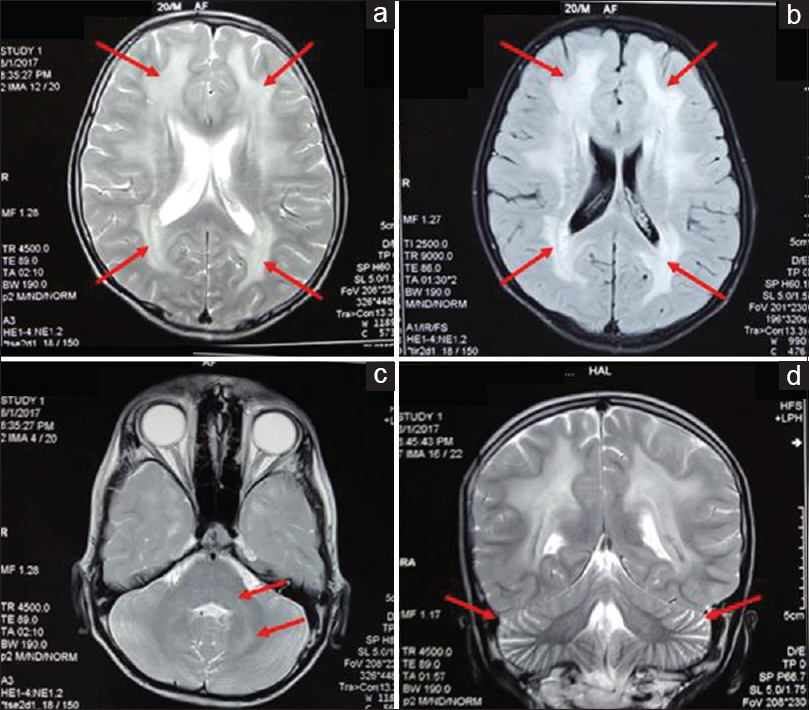
- Axial T2 (a) and fluid-attenuated inversion recovery (b) images showing hyperintensities (arrows) in bilateral frontal and parietal white matter with typical sparing of subcortical “U” fibers. Axial T2 image showing hyperintensities (arrows) in bilateral dentate nuclei, middle cerebellar peduncles, and dorsal pons (c). T2 coronal section showing prominent cerebellar folia (arrows) – suggestive of cerebellar atrophy (d)
After the diagnosis of MNGIE, the possible treatment options were discussed with the parents of the patient. Laparoscopic peritoneal dialysis (PD) catheter placement was done by the surgical gastroenterologist. PD was started after 2 weeks of patient training. He was prescribed one 8 h overnight dwell with 1.5% dextrose of 600 mL volume (30 mL/kg). There were no mechanical complications or peritonitis to date. After 2 months, the patient had improvement in appetite. Postprandial vomiting disappeared completely. Abdominal pain frequency decreased from daily to once or twice a fortnight. His weight increased from 18 to 21.5 kg. Numbness in peripheries vanished completely. However, his mid-arm circumference remained unchanged. Ophthalmoplegia and MRI brain changes remained the same.
Initially, the only one exchange was prescribed; as the patient showed clinical improvement, we advised three exchanges per day. At present three 6 h exchanges of 600 mL, each was being done. The fill volume, however, could not be increased as the tense abdomen could not be tolerated and breathlessness was experienced, probably due to splinting of weak diaphragm and atrophied intercostals and negligible accessory respiratory muscles. We could not test the serum or PD fluid levels of nucleosides. It is not done in any laboratory in India. The patient is under regular monthly follow-up with our facility.
Discussion
MNGIE is a progressive metabolic disorder caused by the cytosolic enzyme thymidine phosphorylase deficiency. TP is coded by TYMP gene (previously ECGF1) which is located on chromosome 22q13.33. TP is seen in almost all human tissues such as gastrointestinal, central, and peripheral nervous system, spleen, liver, urinary bladder, leukocytes, and platelets.[3] TP is absent in kidneys, aorta, and adipose tissue and minimal in muscles. The pathogenesis involves the accumulation of very high concentrations of nucleosides dThd and dUrd along with depletion of nucleotide dCTP.[4] This leads to an imbalance in nucleotide versus nucleoside levels, which results in the destabilization of mtDNA.[5] [Figure 4] This causes interference with mtDNA repair and replication ultimately leading to cell death by apoptosis.

- Pathogenesis of mitochondrial neurogastrointestinal encephalomyopathy
The estimated rate of occurrence is 1–9:1,000,000,[6] and as of 2011, fewer than 200 patients have been described in the medical literature.[7] The natural history of MNGIE is a relentless progression of the disease which invariably culminates in death with severe asthenia and infections being the most common causes of death. Most patients display typical MNGIE features by the age of 20 years.[89] Gastrointestinal symptoms are the earliest to appear.[1] Ocular manifestations and sensory neuropathy in the form of numbness are the second most common manifestations. The treatment includes measures such as fat-soluble vitamin supplementation, levocarnitine, exercise therapy, symptomatic therapy for pain abdomen and vomiting, celiac plexus blockade for severe/recurrent abdominal pain, removal of nucleosides dThd and dUrd by hemodialysis and PD, enzyme replacement therapy, hematopoietic stem cell transplantation (HSCT), and gene therapy.
Only a few patient reports of dialysis as a measure to remove nucleosides had been reported. Spinazzola et al.[10] first reported the hemodialysis in two patients of MNGIE. Patient A underwent one dialysis and patient B underwent three consecutive weekly dialysis treatments. Hemodialysis reduced circulating concentrations of thymidine in both patients. However, the effect was transient. Three hours after dialysis, levels of the nucleoside returned to pretreatment values. Repetitive hemodialysis was also done to maintain basal levels of nucleosides in blood.[11] Yavuz et al.[12] first reported PD in MNGIE. Patient received PD for 3 years. An immediate improvement in gastrointestinal symptoms observed and also weight gain was noted. However, no change in ocular and neurological symptoms had been observed. In another report, the PD was continued for 15 months.[13] An improvement in gastrointestinal symptoms and mitigation of numbness in hands and legs observed; eventually, the patient underwent heterologous HSCT.
In MNGIE, the serum levels of thymidine and deoxyuridine are increased logarithmically and it happens every day. Furthermore, the urinary excretion is saturable in the light of very high serum levels. In this situation, even a small reduction in the serum levels by increasing the clearance by a mere 500 mL/day might allow the imbalance between nucleoside pools to come down, by reversing the reaction catalyzed by TP and favoring the salvage pathway, which allows better mtDNA function in various cell locations.[10]
As this change is not always measurable, this may not reflect as serum level changes yet making clinical improvement. The dissociation between clinical improvement and laboratory measurements had been reported before.[12] This leads us to answer a second logical question that has arisen from the above explanation, how the PD improves neurologic and ophthalmology clinical features notwithstanding the blood–brain barrier to the clearance of nucleosides from cerebrospinal fluid? The explanation stems from the work of Yavuz et al.[12] who had similar observations where there was no demonstrable change in serum levels of nucleosides after a day long PD. The nucleoside concentrations in the dialysate were on average one-third of plasma levels (approximately 100 μmol of both thymidine and deoxyuridine was removed daily by PD). In the same observation, improvement of symptoms occurred in the face of unchanged serum levels of nucleosides which means in addition to high serum nucleoside levels, and pathogenesis of MNGIE involves other yet to be explained mechanisms which might be addressed through PD. In a report published by Sivadasan et al., similar results were seen with resolution of symptoms after starting on PD and return of symptoms when the patient stopped PD which again regressed with PD reinitiation.[14] One proposed mechanism of pathogenesis of MNGIE is that it is the nucleoside accumulation and subsequent reduction of dCTP nucleotides, rather than the deficiency of TP per se, that account for the molecular and phenotypic alterations in MNGIE.[15]
In our patient, the reason for no improvement of ophthalmoplegia and brain changes appeared. Once the neurons of central nervous system (CNS) were damaged, the regrowth was almost unlikely. As glial cells replaced the dead neurons, even if the root cause was corrected, the CNS changes seldom reversed. This was evident in previous observations by Ariaudo et al.[13]
MNGIE is often missed due to its extremely variable presentation from being an acute abdomen to slow relentless wasting disorder and hence needs a very high index of suspicion for diagnosis. A nephrologist has a more promising role in the form of dialysis. Given the limited and difficult treatment options, PD should be considered as a viable and potential treatment option because it obviates the need for vascular access and provides independence to the patient. PD can be used for improving the general condition of the patient when HSCT is anticipated. None of the treatment options available currently have shown to increase the life span of these patients.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain. 2011;134:3326-32.
- [Google Scholar]
- Thymidine phosphorylase mutations cause instability of mitochondrial DNA. Gene. 2005;354:152-6.
- [Google Scholar]
- Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) PLoS Genet. 2011;7:e1002035.
- [Google Scholar]
- Evaluation of gastrointestinal mtDNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) Methods Mol Biol. 2011;755:223-32.
- [Google Scholar]
- Available from: http://www.orpha.net/consor/cgi.bin/index.php
- Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): A consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46:330-7.
- [Google Scholar]
- Mitochondrial neurogastrointestinal encephalomyopathy: An autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol. 2000;47:792-800.
- [Google Scholar]
- Diagnosis and management of MNGIE syndrome in children: Case report and review of the literature. J Pediatr Gastroenterol Nutr. 2002;35:377-83.
- [Google Scholar]
- Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2002;277:4128-33.
- [Google Scholar]
- Pre- and post-dialysis quantitative dosage of thymidine in urine and plasma of a MNGIE patient by using HPLC-ESI-MS/MS. J Mass Spectrom. 2006;41:586-92.
- [Google Scholar]
- Treatment of mitochondrial neurogastrointestinal encephalomyopathy with dialysis. Arch Neurol. 2007;64:435-8.
- [Google Scholar]
- Mitochondrial neurogastrointestinal encephalomyopathy treated with peritoneal dialysis and bone marrow transplantation. J Nephrol. 2015;28:125-7.
- [Google Scholar]
- Pearls and Oy-sters: Mitochondrial neurogastrointestinal encephalomyopathy: Diagnosis and response to peritoneal dialysis. Neurology. 2016;86:e147-50.
- [Google Scholar]
- Mitochondrial neurogastrointestinal encephalomyopathy caused by thymidine phosphorylase enzyme deficiency: From pathogenesis to emerging therapeutic options. Front Cell Neurosci. 2017;11:31.
- [Google Scholar]




