

Translate this page into:
Pedigree Analysis of Polycystic Kidney Disease Patients: Bangladeshi Perspective
Address for correspondence: Dr. Zohora Akther, Department of Anatomy, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh. E-mail: drzohoraakter1982@gmail.com
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background:
Polycystic kidney disease (PKD), an inheritance disorder which is the fourth leading cause of the end-stage renal disease. The inheritance pattern can be diagnosed and confirmed by pedigree analysis. The aim of the present research was to determine the type and frequency of PKD using pedigree analysis.
Materials and Methods:
The present research was designed as a cross-sectional descriptive study. Thirty-eight adult Bangladeshi PKD patients were recruited using a selection checklist from the Department of Nephrology, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh. Data were collected using a data collection sheet after taking informed written consent. The pedigree was drawn using the genetic pedigree chart creation software f-tree V4.0.6. The percentage frequencies of different types of pedigree were calculated using the Statistical Package for the Social Sciences software, version 23.
Results:
A total of 24 (63.20%) had a positive family history and 36.80% (14) had no positive family history. All of the patients with a positive family history had vertical transmission; male and female were equally inheriting the gene. Out of these 24 patients, 4.17% (one), 8.33% (two), and 16.67% (four) had a homozygous/heterozygous state, skip generation, and male to male transmission, respectively.
Conclusions:
Pedigree analysis of PKD patients showed an increased value in early diagnosis and better management and prognosis of the disease.
Keywords
Autosomal dominant polycystic kidney disease (ADPKD)
pedigree
polycystic kidney disease (PKD)

Introduction
Polycystic kidney disease (PKD) is the fourth leading cause of end-stage renal disease (ESRD).[1] It is found in all races and occurs equally in men and women.[2]
This genetic disease is transmitted from parents to children.[2] This inherited disease is caused by mutated genes that create a specific aberrant protein that causes disruption of tubule formation.[3] Of the various cystic diseases, autosomal dominant PKD (ADPKD) is the most prevalent hereditary kidney disease with an incidence of 1 in 400–1 in 1000.[4,5] The inheritance pattern can be identified through pedigree analysis and can helps in the identification of patients and families who are at higher risk for genetic illnesses, as well as the optimization of counseling, screening, and diagnostic testing with the objective of disease prevention or early detection and management.[6]
When constructing a pedigree, it is essential to be methodical and use internationally recognized symbols and patterns.[7,8]
Diagnosis of PKD is based on clinical features and renal ultrasonographic (USG) findings in Bangladesh. But it is the demand of time to identify the inheritance pattern of PKD with the help of pedigree along with USG reports and clinical features like in other countries. The aim of the present research is to determine the types and frequencies of PKD through pedigree analysis.
Materials and Methods
The present research was designed as a cross-sectional descriptive study. The research was carried out in the Department of Anatomy, Bangabandhu Sheikh Mujib Medical University (BSMMU), Dhaka, from March 2021 to February 2022, after getting formal approval from the Institutional Review Board (IRB) of BSMMU. Thirty-eight patients were selected using the “selection checklist” and confirmed by an assigned nephrologist based on USG reports and clinical features. After obtaining an informed consent, the data collection sheet was filled out to evaluate the personal and family histories of the patients. Sometimes, telephone calls were made to obtain additional information.
History was taken meticulously regarding maternal and paternal families for three to five generations using a data collection sheet. The necessary information was entered into the genetic pedigree chart creation software f-tree V4.0.6 and the pedigree was generated automatically in the software. The data were analyzed using the Statistical Package for the Social Sciences (SPSS) software, version 23. The percentage frequency of different types of pedigree was calculated using SPSS.
Results
All participants (100%, 38) were diagnosed as ADPKD by an assigned nephrologist based on clinical features and USG reports. The mean age of the ADPKD patients was 37.76±11.88 and 34.2% patients were 37 to 46 years old [Table 1].
| Age range (years) | Percentage frequency (total number) | Mean±SD (years) |
|---|---|---|
| 17-26 | 21.1 (8) | |
| 27-36 | 23.7 (9) | |
| 37-46 | 34.2 (13) | 37.76±11.88 |
| 47-56 | 13.2 (5) | |
| 57-66 | 7.9 (3) |
ADPKD=Autosomal dominant polycystic kidney disease, SD=Standard deviation
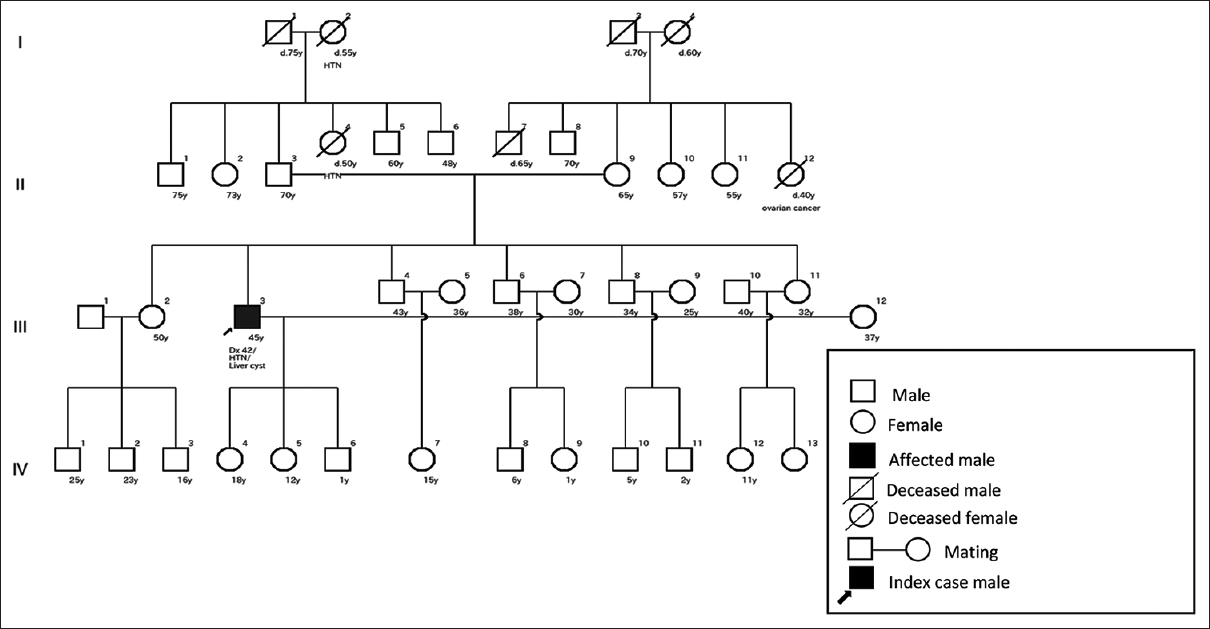
Among 38 clinically diagnosed ADPKD patients, 63.20% (24) had a positive family history. All these 24 patients had vertical transmission; male and female were equally inheriting the gene and an abnormal gene from one parent can cause the disease, as shown in Figure 1. Regarding zygosity, 4.17% (one) patients had a homozygous/heterozygous state, as shown in Figure 1. Male to male transmission was present in 16.67% (four) patients, as shown in Figure 1.

- Pedigree of an ADPKD patient. Note the presence of vertical transmission, homozygote/heterozygote state, male to male transmission
A total of 14 (36.80%) had no positive family history. Only the affected person came as an index case. Among those with no positive family history, 42.86% (six) ADPKD patients had hypertensive parents. Beside this, only 7.14% (one) siblings and 7.14% (one) parents had a liver disease, as shown in Figure 2. The remaining 42.86% (six) patients had no renal or extrarenal feature in the family related to ADPKD, as shown in Figure 3.
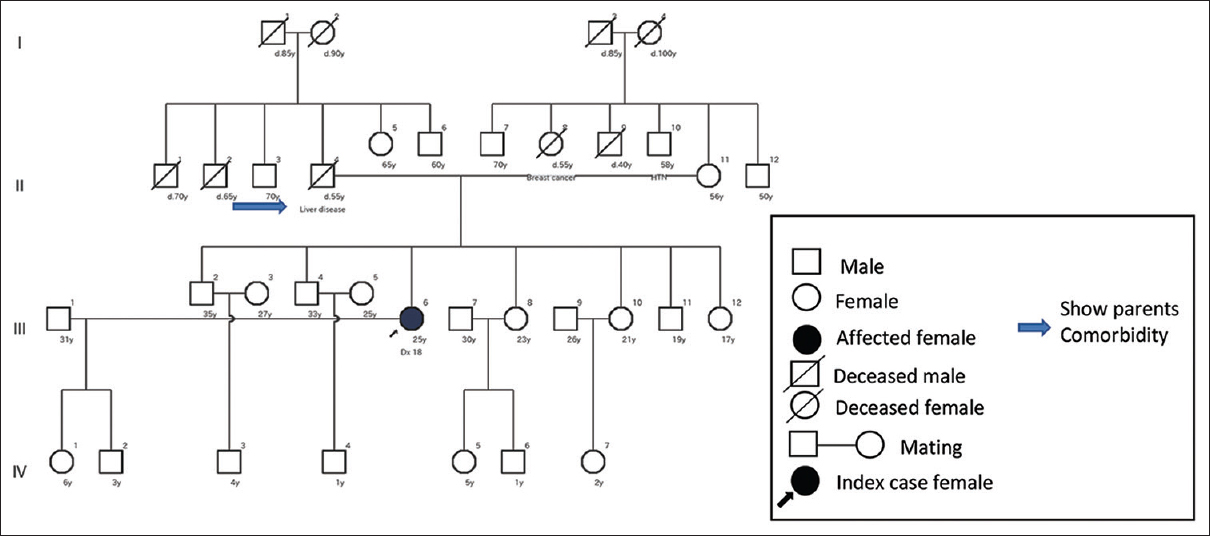
- Pedigree of an ADPKD patient with no evidence of positive family history. Note the parent’s comorbidity (father)
- Pedigree of an ADPKD patient with no evidence of positive family history. Note no renal or extrarenal features present in the family
USG reports showed 78.95% (30) patients had bilaterally enlarged kidneys. Multiple cysts in both kidneys were found in 86.84% (33) patients, as shown in Table 2. Hypertension (HTN) was found to be in a higher percentage (78.90%) among the 38 ADPKD patients. Other renal and extrarenal comorbidities of ADPKD patients are shown in Table 3.
| Criterion | Percentage frequency (total number) |
|---|---|
| Enlarged kidney | |
| Bilaterally enlarged kidneys | 78.95 (30) |
| Unilaterally enlarged kidney | 10.53 (4) |
| Normal-sized kidney | 10.53 (4) |
| Cyst | |
| Multiple cysts in both kidneys | 86.84 (33) |
| Hepatic cyst | 13.20 (5) |
ADPKD=Autosomal dominant polycystic kidney disease
| Comorbidity | Percentage frequency (total number) |
|---|---|
| Renal | |
| Hypertension | 78.90 (30) |
| Nephrolithiasis | 13.20 (5) |
| Renal failure | 2.60 (1) |
| Extrarenal | |
| Hepatic cyst | 13.20 (5) |
| Heart disease | 5.3 (2) |
| Pancreatic cyst | 0 |
| Seminal vesical cyst | 0 |
| Cerebral aneurism | 0 |
ADPKD=Autosomal dominant polycystic kidney disease
Discussion
Positive family history with vertical transmission is one of the criteria of autosomal dominant diseases.[9] As it is a dominant disease, the present research found 63.20% of patients met this criterion. A cohort study of the Asian Indian population found 61.6% of patients have a positive family history, which supports the present research.[11] In a dominant disease, both male and female members have an equal chance to become affected. If one parent has the affected gene, it could affect their offspring.[10] Patients with a positive family history in the present study met this criterion, as shown in Figure 1. If all the offspring become affected from a single affected parent, the affected parents may be in a homozygote or heterozygote state, as shown in Figure 1. In a dominant disease, homozygous individuals become severely affected or have an early age of onset.[9] In the present research, the affected individual was diagnosed in their fourth decade of life and the renal function did not show too much decline. These suggest the affected individual may be heterozygous. In the present research, 16.67% of patients showed male to male transmission, which is another criterion for autosomal dominant disease, as shown in Figure 1.[9]
Among the patients of ADPKD with no evidence of positive family history, parents had comorbidities like HTN or a history of stroke (may be due to cerebral aneurysm), liver disease, and 7.14% siblings had a liver disease. These features suggest that chance of reduces penetrance. Incomplete or reduced penetrance shows that some individuals fail to express the trait, even though they contain the allele.[12] These features are also in favor of a dominant disease. In another 42.86% of patients with no evidence of positive family history, the parents and siblings had no renal or extrarenal comorbidity, which may suggest chances of de novo mutation of the index case. Audrézet et al.[13] reported that there is a high frequency of de novo mutations in case of ADPKD in the European population and it was confirmed by molecular analysis of the responsible genes. Another study on North American population revealed that 50% of ADPKD patients have de novo mutation, which was further confirmed by parents’ genetic analysis.[14] Besides these, a study on Asian Indian population showed that 38.4% of ADPKD patients had no positive family history.[11] On the other hand, mistakes during DNA replication can result in de novo mutations due to improper nucleotide incorporation by DNA polymerases. DNA lesions can also emerge as a consequence of exogenous or endogenous mutagens like ionizing radiation, DNA-reactive compounds, and reactive oxygen species.[15] In the present research, none of the patients had any history of occupational exposure.
In the present study, 78.95% of patients had bilaterally enlarged kidneys and multiple cysts were found in both kidneys in 86.84% of patients, which is comparable to another Bangladeshi study where bilaterally enlarged kidneys and multiple cysts in both kidneys were found in 70% and 90% of patients, respectively.[16] Another study also reported excessively enlarged kidneys due to formation and enlargement of numerous cysts.[17]
Conclusions
Diagnosis of PKD with pedigree analysis shows value of genetic counseling followed by decision-making, early diagnosis, and better management and prognosis of the disease.
Financial support and sponsorship
The research was funded by BSMMU thesis grant and self.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
The authors of this article thank the Department of Anatomy and the Department of Nephrology, Bangabandhu Sheikh Mujib Medical University (BSMMU), for providing the infrastructure and other research facilities. They also express their gratitude to the participants of the research.
References
- Autosomal dominant polycystic kidney disease:Core curriculum 2016. Am J Kidney Dis. 2016;67:792-810.
- [Google Scholar]
- Polycystic kidney disease | National Kidney Foundation. National Kidney Foundation; 2018. p. :1.
- Identification of novel PKD1 and PKD2 mutations in Korean patients with autosomal dominant polycystic kidney disease. BMC Med Genet. 2014;15:129.
- [Google Scholar]
- Novel mutations in the PKD1 and PKD2 genes of Chinese patients with autosomal dominant polycystic kidney disease. Kidney Blood Press Res. 2018;43:297-309.
- [Google Scholar]
- Family history screening:Use of the three generation pedigree in clinical practice. J Obstet Gynaecol Canada.. 2010;32:663-72.
- [Google Scholar]
- Perceptions of family history and genetic testing and feasibility of pedigree development among African Americans with hypertension. Eur J Cardiovasc Nurs. 2015;14:8-15.
- [Google Scholar]
- Importance of pedigree in patients with familial epilepsy and intellectual disability. Sudan J Paediatr. 2019;19:52-6.
- [Google Scholar]
- Emery's Elements of Medical Genetics (15th ed). New York: Elsevier; 2017.
- Principles of Genetics (5th ed). New Jersey: John Wiley and Sons; 2010.
- Identification of PKD1 and PKD2 gene variants in a cohort of 125 Asian Indian patients of ADPKD. J Hum Genet. 2019;64:409-19.
- [Google Scholar]
- Reduced penetrance in human inherited disease. Egypt J Med Hum Genet. 2014;15:103-11.
- [Google Scholar]
- Autosomal dominant polycystic kidney disease:Comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat. 2012;33:1239-50.
- [Google Scholar]
- Polycystic kidney disease without an apparent family history. J Am Soc Nephrol. 2017;28:2768-76.
- [Google Scholar]
- New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016;17:1-19.
- [Google Scholar]
- Clinical features of autosomal dominant polycystic kidney disease (ADPKD) among bangladeshi patients in a tertiary care center. Int J Med Res Prof. 2019;5:109-11.
- [Google Scholar]
- Demographic, diagnostic and therapeutic characteristics of autosomal dominant polycystic kidney disease in Ghana. BMC Nephrol. 2021;22:1-8.
- [Google Scholar]




