

Translate this page into:
Phenotype Spectrum in Tunisian Population with NPHP1 Deletion
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Introduction:
Nephronophthisis (NPHP) is a tubulointerstitial kidney disorder with an autosomal recessive inheritance pattern. Its genetic heterogeneity contributes to phenotype variability. The most frequent etiology of juvenile nephronophthisis is a mutation in the nephronophthisis type 1 (NPHP1) gene. This study aimed to evaluate the genotype-phenotype correlation in NPHP1 gene mutation.
Methods:
A multicenter retrospective study was performed over 20 years from 1998 to 2018 to describe the clinical, biological, and radiological features associated with the large deletion NPHP1 gene in 32 patients.
Results:
The incidence of NPHP1 was 1.6/204041. Eighty-one percent of our patients were born out of consanguineous marriages. The mean age at diagnosis was 14 ± 7 years. The patients were divided into three groups: isolated nephronophthisis (72%), syndromic nephronophthisis (19%), and patients without recognizable syndrome (9%). Intrafamilial and geographical variability was observed in syndrome diagnoses and in age at the onset of CKD stage 5. Genotype frequency varied between 50% and 100% in genealogical data. Juvenile (47%), adolescent (37%), and adult (13%) clinical forms have been distinguished by the onset of CKD stage 5. The five-year survival rate of renal transplantation was 80%.
Conclusion:
Given the broad clinical spectrum of NPHP1 associated with the large deletion of the NPHP1 gene, no genotype-phenotype correlation could be established.
Keywords
Chronic kidney disease
ciliopathy
nephronophthisis
NPHP1
renal replacement therapy
renal transplantation

Introduction
Nephronophthisis (NPHP) is an autosomal recessive renal ciliopathy causing cystic kidney disease, renal fibrosis, and end-stage renal failure. It is the most common genetic cause of chronic kidney disease (CKD) stage 5 in the first three decades of life.[1] To date, 25 NPHP genes have been identified.[2] Nephronophthisis 1 gene mutation that leads to nephronophthisis 1 (NPHP1: OMIM 256100) is yet the most frequent genetic cause and accounts for almost 21% of NPHP cases.[3]
The NPHP1 gene was mutated in 30%–85% of patients with juvenile NPHP. The major NPHP1 gene defect is a homozygous large deletion.[4,5] Furthermore, gene products interact with the protein modules of nephrocystins and share expression in centrosomes and primary cilia. This results in impaired ciliary structure and function. Nephrocystins are ubiquitous proteins expressed in kidneys and several tissues (retinitis, central nervous system, liver, bones, and genital organs), which might explain why organs than the kidneys can also be affected. Extrarenal organ disorders define NPHP-related syndromes (Joubert syndrome, Senior Loken syndrome, Cogan syndrome, Saldino–Mainzer syndrome, and Boichis syndrome).[6]
Several studies attempted to establish correlations between each phenotype observed and underlying NPHP1 gene mutation.[7-9] No clear genotype-phenotype correlation has been defined.[2,10]
To describe the broad phenotypic spectrum related to the NPHP1 gene mutation in the Tunisian population and to evaluate the genotype–phenotype correlation, we retrospectively reviewed a multicenter Tunisian cohort over 20 years (1998–2018).
Patients and Methods
Study cohort
We included all the patients with a large deletion of the NPHP1 gene at from six different pediatric and nephrology across the country. The deletion was defined as a lack of amplification products of all three NPHP exons (exons 2, 14, and 19).
To analyze the phenotype–genotype correlation, we distinguished three groups of patients based on clinical features: isolated NPHP1, syndromic NPHP1, and those with extrarenal features that do not constitute a recognizable syndrome.
All individuals were screened for homozygous NPHP1 deletions by using polymerase chain reaction (PCR)-based gel electrophoresis. PCR for mutation analysis was performed in two laboratories at Charles Nicolle and Sahloul hospitals. The blood sample in order to extract the DNA of the patients and their families was obtained after informed and signed consent. Three pairs of primers amplifying three different exons of the NPHP1 gene (exons 2, 14, and 19) were PCR amplified. The lack of amplification products of all three NPHP exons was considered a homozygous deletion in NPHP1.
Clinical assessment and definitions of kidney function were determined using the eGFR according to the Schwartz formula for pediatric patients[11] and the MDRD formula for adults.[12] We defined CKD stages according to the KDIGO classification.[13] We expressed a measure of maximum kidney length as means ± SD according to the height of patients.
Statistical analysis
We realized data analysis by using the software program statistical package for social science (SPSS) version 20. Descriptive statistics used comprised percentages and mean ± standard deviation (SD) and median. The level of statistical significance was predefined as P < 0,05. We performed the comparison of data of patients with isolated renal features versus patients with extrarenal manifestations by using the Chi-square test.
Results
Thirty-two patients from 25 families were recruited. The incidence of NPHP1 in our study was 0.78/100000 births. There were 25 sporadic cases and five family forms. Sex distribution showed a ratio of 1.3 (14 females and 18 males). Parental consanguinity was found in 26 patients (81%). The main age at diagnosis was 14 ± 7 years (range: 4–33). It was significantly delayed from age of onset of symptoms with an average of 50 months (P = 0.004).
Clinical presentation
At clinical presentation, uremic symptoms with vomiting, nausea, and anorexia, were reported in 24% and growth failure in 13%. Joint pain led to a diagnosis of severe renal failure 6 months to 3 years later in the disease course. Polyuria and polydipsia were reported in 13% and 8% of cases, respectively.
Reduced urinary concentrating capacity was detected in all patients. Proteinuria was found in three patients, exclusively in CKD stage 5. Hematuria was reported in two cases. Hypertension was detected in five, all with advanced CKD. A deformity of lower limbs was detected in four patients with CKD stage 5. Dysmorphic features were observed in two patients. Flat occiput, broad forehead hypodontia, and clinodactyly were associated in patient 1. Patient 2 had scaphocephaly, hypertelorism, webbed neck, and brachydactyly.
Visual impairment was reported in 25% of patients (8/32), including nystagmus (n = 4), high myopia (n = 3), strabismus (n = 3), amblyopia (n = 1), and cataract (n = 1). The fundoscopic examination was performed in 27/32 patients and revealed retinitis pigmentasum (RP) in five cases. Table 1 lists the eye abnormalities [Table 1]. Electroretinogram analysis denoted a decreased cone and rod response.
| Case | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| High myopia | + | - | + | + | - |
| Amblyopia | - | - | - | + | - |
| Strabismus | + | + | - | - | - |
| Nystagmus | - | + | - | + | - |
| Choroidal atrophy | Localized | Diffuse, with thin vessels | Diffuse | ||
| ERG* response | + | + | + |
ERG=Electroretinogram
We documented neurological manifestations in 22% of cases (7/32), including mental retardation (n = 2), microcephaly (n = 1), oculomotor apraxia (n = 1), ataxia (n = 1), and neurosensitive defect (n = 1). Magnetic resonance imaging (MRI) performed in four patients (12%) showed typical cerebellar vermis hypoplasia in one [Figure 1].

- Hypoplasia of vermix (sign of the molar) associated with NPHP1
Overall, 23 of 32 patients (72%) presented with isolated NPHP1. Six patients exhibited NPHP1-related ciliopathies, namely Joubert syndrome (n = 1), Senior–Lœken syndrome (n = 4), and congenital oculomotor apraxia (n = 2), as described in Table 2.
| Case | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Age of diagnosis | 13 | 9 | 5 | 9 | 11 | 11 |
| Physical examination | Microcephaly | Mental retardation | Oculomotor apraxia | Mental retardation | - | - |
| - | Nystagmus | Nystagmus | Nystagmus | Nystagmus | - | |
| Strabismus | Strabismus | Strabismus | Amblyopia | |||
| High myopia | High myopia | |||||
| BER | Transmission deafness | Perception deafness | - | Perception deafness | ||
| Fundoscopic examination | RP | RP | - | RP | RP | RP |
| MRI | N | N | N | Vermis hypoplasia | N | N/D |
| NPH-related syndromes | SLS | SLS | Cogan syndrome | Joubert syndrome | SLS | SLS |
BER=brainstem evoked response, RP=Retinitis pigmentasum, MRI=magnetic resonanace imaging, N=Normal, N/D=Not done, SLS=Senior Loken Syndrome, NPH=nephronophtisis
RP with congenital deafness was observed in three patients (1, 4, and 6) and was associated with moderate cerebral vermis hypoplasia and mental retardation in patient 4 [Table 2].
For those with extrarenal features (n = 3) that do not constitute a recognizable syndrome, we observed electromyography confirmed neurosensitive defect, isolated mental retardation with normal MRI, and an association of ataxia/nystagmus with undone MRI.
At the renal ultrasound, seven out of 32 patients (23%) had normal-sized kidneys, while 25 patients had small kidneys (<−2 SD). Increased echogenicity (44%) was detected in all stages of CKD.
In the isolated NPHP1 group, eight out of 21 patients presented with renal cysts compared with two out of six in the NPHP1 syndromic form. No significant association was found between cyst presence and NPHP1 form (P = 0.470). Liver involvement was not reported in our cohort.
Renal biopsy
Renal biopsy was performed in 5/32 patients. It showed glomeruli with segmental sclerosis and periglomerular fibrosis compatible with chronic tubulointerstitial nephritis.
Kidney function
The average of serum creatinine at the time of diagnosis of NPHP1 was 396.9 μmol/L (range77.5-1222 μmol/L). Five patients (15%) had a CKD stage 3 and 16 had a stage 4. After 1 year of evolution, 19 patients evolved to CKD stage 5. The average follow-up time was 9 years. For the five remaining after exclusion of patients who had a kidney transplant and those who were insufficiently followed, two preserved stable kidney function so far, while three exhibited significantly improved estimated glomerular filtration rate. Overall, 94% of patients developed CKD stage 5 during the follow-up. The age of reaching an end-stage renal disease (ESRD) was variable with a minimum of 6 years and a maximum of 33 years. Within our NPHP1 cohort, 14 children (47%) presented juvenile NPHP1 with CKD stage 5 before 15 years old. Eleven (37%) were adolescents with a mean age of ESRD of 19. Five adults (16%) reached ESRD beyond 25 years old. We did not observe any infantile form. All patients with ESRD received dialysis.
No significant difference was found in the kidney survival with non-syndromic NPHP1 and those with a syndromic form (P = 0.512) [Figure 2].
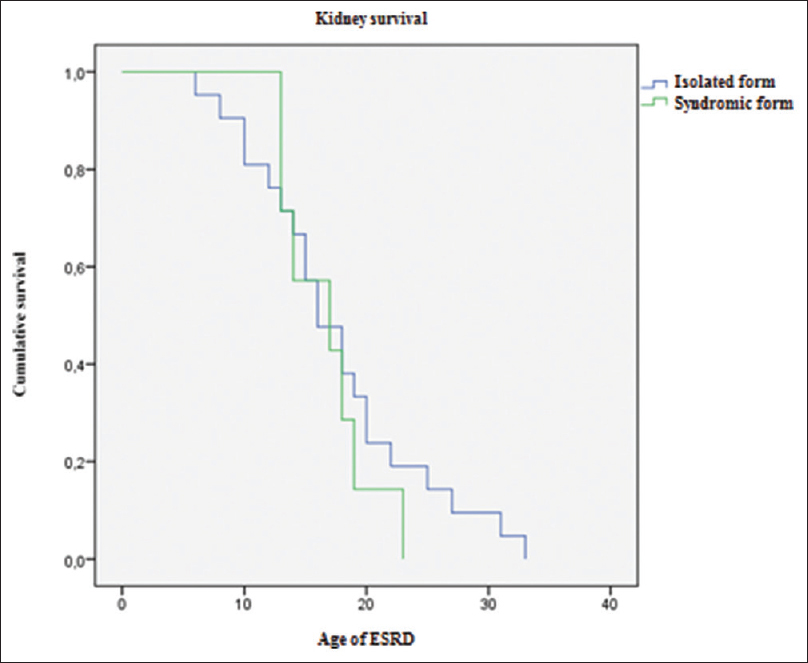
- Kaplan–Meier analysis of kidney survival among NPHP1 isolated and syndromic forms
Intrafamilial variability
We identified five families with consanguinity. Three of them had a non-syndromic form F1, F2, F3 and NPHP1 syndromic form in families F4 and F5. The two affected siblings in F5 showed similar syndromes. We observed that the siblings in F4 had different clinical manifestations.
Kidney transplant
Twelve out of 32 (37%) had undergone a kidney transplant One of them received a kidney transplant prior to the diagnosis of NPHP1. The mean age at transplant was 19 ± 7 years.
Following transplantation, mean serum creatinine was 78.6 μmol/L at 1 year and 54 μmol/L at 5 years. We reported two graft failures out of 12 transplant patients.
One of them lost his graft in the seventh-year post-transplant due to chronic allograft dysfunction Graft survival was 80% at 5 years and 67% at 10 years. We do not note the recurrence of nephronophthisis in our cohort.
Discussion
In this study, we estimated pathogenic allele frequency as 1.15%. Consanguinity was found among 81% of patients. The mean age at diagnosis was 14 ± 7 years. According to the presence or absence of extrarenal features, patients were classified into patients with non-syndromic nephronophthisis (72%), syndromic form (19%), and patients with nephronophthisis and extrarenal features not constituting a recognizable syndrome (9%). While investigating renal phenotype in isolated and syndromic forms, no significant differences were found in kidney sizes, renal cysts), and main age at the onset of CKD stage 5.
The 20-year observation period of our study made his originality. Thus, it allowed us to establish a temporal sequence with enough hindsight to study the evolution of the disease over time. In contrast, carrying out a multicenter study allows the determination of the epidemiological parameters and limits the sampling fluctuation.
The incidence of NPHP1 in our study is remarkably lower compared to reports in Finland (1.3/100,000 births), Canada (1/50,000 births), and Europe (1/61,800 births).[14-16] However, those statistics were reported 20 years ago and included all types of NPHP. To the best of our knowledge, no recent data on disease incidence is available.
Our finding was consistent with a prior study that established that the Tunisian population is characterized by a high incidence of hereditary nephropathies, which accounts for 31.3% of identified causes of CKD stage 5 following congenital anomalies of the kidneys and urinary tract that mainly leads to CKD.[14] This proportion is higher than that reported in international registries in which the percentage of hereditary nephropathies ranged between 2% and 22% following congenital anomalies of the kidneys and urinary tract and glomerulonephritis.[15] This difference may be explained by a higher rate of consanguineous marriages in Tunisia.[17]
In our finding, patients were diagnosed at an older age in comparison with the Egyptian cohort aged mainly 9–10 years old at first diagnosis.[18] Polyuria and polydipsia were the early symptoms of the disease, usually occurring at around 6 years.[15] However, they accounted for only 21% of the primary reasons for consultation in our study.
To our knowledge, no specific dysmorphic features have been associated with NPHP1. Typical facial appearance has been described in Joubert syndrome, including a broad forehead, ocular hypertelorism, and polydactyly.[19]
Conversely, we found hypertelorism in an isolated NPHP1 form and broad forehead in Cogan syndrome associated with atypical features in each patient. Some extremity abnormalities such as clinodactyly and brachydactyly had not been described before. Visual impairment was the most common extrarenal manifestation in our study, which is in line with the worldwide cohort of Caridi and Stokman series.[20,21] Neurologic symptoms were present more frequently than in respective reports [Table 3]. In our study, we found pathognomonic features of Joubert syndrome in one case and Senior Loken syndrome in four cases with NPHP1 deletion. These syndromes are known with other nephrocystin mutations (NPHP5, 6, and 8), but the NPHP1 mutation associated with these syndromes has been described in the literature.[22]
| Our study (n=32), 2018 | König et al.[23] Germany (n=60), 2016 | Caridi et al.[20] Italy (n=56), 2006 | Stokman et al.[21] Netherland, (n=16), 2018 | |
|---|---|---|---|---|
| Neurological manifestations | 22% | 25% | 8,9% | 7% |
| Cognitive defect | 9% | 19% | 3.5% | 7% |
| Microcephaly | 3% | - | - | |
| Oculomotor apraxia | 3% | 10% | 3.5% | 7% |
| Ataxia | 3% | 7% | 7.1% | - |
| Neurosensitif defect | 3% | 0% | 0% | - |
| Vermis hypoplasia | 3% | 3% | 3.3% |
All patients had reduced eGFR at the time of diagnosis. The progression of NPHP1 to renal failure is inevitable.[16] The predominance of severe and terminal CKD is indicative of the delayed diagnosis of the disease. The categorization into three groups (isolated NPHP1, syndromic NPHP1, and non-syndromic NPHP1) in our cohort, as done in the study by Stokman, helped us to improve the analysis of the evolution of kidney function in the different categories.[21] Stokman reported the presence of CKD stage 5 on admission in 78% of isolated forms and 50% of syndromic forms[21] versus 39% and 14%, respectively, in our study. The severe stage was the majority in our study with no significant difference in renal survival between the two forms. The average alkaline reserve was 18.61 mmol/L. Metabolic acidosis is explained by advanced renal failure at the time of diagnosis. It is implicated in the failure to thrive, as observed in most of the studies.
Nephrocystin-1 has been localized at the cilia connector and outer segment of photoreceptors where it interacts with other proteins mutated in RP (RP3-RPGR-RPGRIP1), explaining the ocular phenotype associated with NPHP1. Its interaction with jouberin, a product of the AHI1 gene, on the same signaling pathway, has been mainly implicated in the associated neurological manifestations.
However, the mutation studied, being identical in all individuals, would not alone explain the variability of the observed phenotypic spectrum. Factors other than the mutation itself would be involved in the correlation between the genotype and phenotype of NPHP1.[24]
Variability of clinical expression between members of the same family has long suggested the involvement of non-genetic factors. Phenotype divergence has been reported in monozygotic twins, thus carrying the same mutation of the NPHP1 gene.[25] In contrast, the pleiotropy in NPHP1 can explain the fact that we had Senio–Loken and Joubert syndromes with nephrocystin mutation different than those described in the literature.[22] Epigenetic and environmental factors are thought to be involved in disease expression.[26]
Gene modifiers can determine the expression of the phenotype and the age of onset of CKD stage 5.[27] Its variability within siblings implies the epistatic expression of the NPHP6 and AH1 modifier genes, associated with added mutations.[28]
The predominance of associated forms in the central part of the country and the concentration of adult forms in the north-western regions reinforces the hypothesis of the involvement of environmental factors in the transmission of the disease.
NPHP accounts for 2.8% of kidney transplants according to the NAPRTCS registry established in 2006.[29] In Tunisia, as we do not have a national registry of kidney diseases, this one is necessary. Among the study participants, 37% had been kidney transplanted. The shortage of organs is the main cause of low access to transplantation.[14]
The average age of kidney transplantation in our study was 18.5 years. This is late in comparison with a mean age of transplantation of 12 years ± 4.1 reported in other series of NPHP (n = 17).[30] Acute rejection was reported in 11% of NPHP cases.[29] It was observed in 8% of NPHP cases in our study.
Chronic rejection was reported in 5.5% of NPHP cases (n = 249).[28] Viral and bacterial graft infection was reported in 0.9%. The recurrence of the initial disease has never been described.
The observed decline in renal function was moderate with mean clearance at 1, 3, and 5 years being 78.6, 76.4, and 54 mL/min/m2, respectively. These figures are lower than those reported by Tayfur et al., where creatinine clearance in post-renal transplantation in a patient with NPHP was estimated to be 85, 75.2, and 83.2 mL/min/1.73 m2, respectively (n = 9).[31]
Pre-emptive transplantation remains the optimal treatment for improved graft life expectancy before the onset of irreversible clinical signs of CKD.
Some parameters of our study could have been studied more precisely by a prospective study, allowing a direct and precise calculation of the incidence rate of the disease and a selection of patients at an early stage. In addition, a larger sampling would have allowed a more rigorous data collection.
Conclusion
At the end of this study and given the broad clinical spectrum of NPHP1 associated with the large deletion of the NPHP1 gene, we can conclude that the genotype–phenotype correlation cannot be established. The early molecular confirmation of this disease allows appropriate therapeutic management, in particular pre-emptive kidney transplantation. Adequate genetic counseling must be given and prenatal diagnosis can be offered to affected families.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We would like to thank all the staff of the pediatric department and the genetic department at Charles Nicolle Hospital for their support of clinical practice. We also would like to thank patients and their guardians for their providing clinical data for this study.
References
- Nephronophthisis:Disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20:23-35.
- [Google Scholar]
- Nephronophthisis:A review of genotype-phenotype correlation. Nephrology. 2018;23:904-11.
- [Google Scholar]
- Clinical and molecular heterogeneity of juvenile nephronophthisis in Italy:Insights from molecular screening. Am J Kidney Dis. 2000;35:44-51.
- [Google Scholar]
- Nephronophthisis and autosomal dominant interstitial kidney disease (ADIKD) Pediatr Kidney Dis 2016:369-88.
- [Google Scholar]
- Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132:865-84.
- [Google Scholar]
- Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy. Exp Mol Med. 2016;48:e251.
- [Google Scholar]
- Many genes—One disease?Genetics of nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr. 2018;5:287.
- [Google Scholar]
- Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 2011;80:1239-45.
- [Google Scholar]
- New equations to estimate GFR in children with CKD. J Am Soc Nephrol. 2009;20:629-37.
- [Google Scholar]
- Definition and classification of chronic kidney disease:A position statement from Kidney Disease:Improving Global Outcomes (KDIGO) Kidney Int. 2005;67:2089-100.
- [Google Scholar]
- KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120:c179-84.
- [Google Scholar]
- Épidémiologie de l'insuffisance rénale terminale de l'enfant en Tunisie [Etiologies of end-stage renal disease of children in Tunisia. Nephrol Ther. 2016;12:166-70.
- [Google Scholar]
- Epidemiology of chronic kidney disease in children. Pediatr Nephrol. 2012;27:363-73.
- [Google Scholar]
- Differential impact of consanguineous marriages on autosomal recessive diseases in Tunisia. Am J Hum Biol. 2016;28:171-80.
- [Google Scholar]
- Clinical characterization and NPHP1 mutations in nephronophthisis and associated ciliopathies:A single center experience. Saudi J Kidney Dis Transpl. 2012;23:1090-8.
- [Google Scholar]
- Joubert syndrome (and related disorders) (OMIM 213300) Eur J Hum Genet. 2007;15:511-21.
- [Google Scholar]
- Nephronophthisis type 1 deletion syndrome with neurological symptoms:Prevalence and significance of the association. Kidney Int. 2006;70:1342-7.
- [Google Scholar]
- Clinical and genetic analyses of a Dutch cohort of 40 patients with a nephronophthisis-related ciliopathy. Pediatr Nephrol. 2018;33:1701-12.
- [Google Scholar]
- Stokman M, ed. Nephronophthisis. Seattle: GeneReviews; 2016.
- Phenotypic spectrum of children with nephronophthisis and related ciliopathies. Clin J Am Soc Nephrol. 2017;12:1974-83.
- [Google Scholar]
- Juvenile nephronophthisis and dysthyroidism:A rare association. CEN Case Rep. 2017;6:98-104.
- [Google Scholar]
- Joubert syndrome:Monozygotic twins with discordant phenotypes. J Child Neurol. 1999;14:649-54.
- [Google Scholar]
- Epigenetics of discordant monozygotic twins:Implications for disease. Genome Med. 2014;6:60.
- [Google Scholar]
- NPHP1 (Nephrocystin-1) Gene deletions cause adult-onset ESRD. J Am Soc Nephrol. 2018;29:1772-9.
- [Google Scholar]
- High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis:Potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18:1566-75.
- [Google Scholar]
- Outcomes of kidney transplantation in children with nephronophthisis:An analysis of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Registry. Pediatr Transplant. 2008;12:878-82.
- [Google Scholar]
- Our experience of patients with juvenile nephronophthisis after renal transplantation. Transplantation. 2018;102:S297.
- [Google Scholar]
- Follow-up of patients with juvenile nephronophthisis after renal transplantation:A single center experience. Transplant Proc. 2011;43:847-9.
- [Google Scholar]




