

Translate this page into:
Rare, Yet Emerging Cause of Graft Dysfunction—ALECT 2 Amyloidosis
Address for correspondence: Dr. Kulwant Singh, Division of Nephrology, Grecian Hospital, Sector 69, Mohali - 160 062, Punjab, India. E-mail: kulmin2k2@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Amyloidosis is characterized by pathological deposition of abnormal protein aggregates in various tissues, AL protein being the commonest. Amyloidosis derived from leukocyte cell-derived chemotaxin 2 (LECT2) is a recently recognized form of amyloidosis in the United States with predominant involvement of kidney and liver. We present a case of ALECT2 renal amyloid in a transplant recipient who presented with gradual worsening of graft function and subnephrotic proteinuria. To our knowledge, this is first case of LECT2 amyloidosis from Northern India in a transplant recipient. There is no effective therapy for amyloidosis derived from leukocyte cell-derived chemotaxin 2.
Keywords
ALECT2 amyloid
graft dysfunction
renal transplant

Introduction
Amyloidosis is characterized by extracellular deposition of insoluble fibers that result from abnormal protein folding. AL amyloidosis is most frequent type of amyloidosis in United States, whereas in developing countries AA amyloidosis is more common.[1] In 2008, Benson et al. discovered a new form of amyloid derived from leukocyte cell-derived chemotaxin 2 (ALECT2) in a nephrectomy specimen done for renal cell carcinoma.[2] ALECT 2 amyloidosis is now as common as AA amyloidosis in United States accounting for 2.7–10% of patients with renal amyloidosis.[3] ALECT2 amyloidosis exhibits a strong ethnic bias, with 88–92% of reported patients being Hispanics mostly of Mexican descent.[4] We describe a case of renal ALECT2 amyloidosis in a renal transplant recipient who presented with gradual renal dysfunction and subnephrotic proteinuria. This case report highlights the fact that primary amyloidosis is one of the underdiagnosed causes of renal dysfunction.
Case Presentation
A forty-three-year-old renal allograft recipient of Indo Aryan lineage was referred for evaluation of gradual rise in serum creatinine. He was diagnosed with advanced renal dysfunction 18 years back when he presented with breathlessness and accelerated blood pressure. Evaluation revealed advanced azotemia, subnephrotic proteinuria, and bilateral shrunken kidneys. Renal biopsy was not done in view of shrunken kidneys. He underwent first renal transplant in 2001 with mother as donor, with no induction and maintenance with cyclosporine, azathioprine, and prednisolone. He had acute graft dysfunction after 6 months of transplant and developed graft loss due to refractory cell-mediated rejection. He underwent second transplant in 2003 with father as donor (2 years after the first transplant). No induction was given and he was maintained on tacrolimus, azathioprine, and prednisolone. His serum creatinine remained in the range of 1.2–1.4 mg/dl for next 12 years. In 2017, his creatinine rose to 4.6 mg/dl and biopsy revealed cell-mediated rejection with evidence of chronic calcineurin inhibitor toxicity. He was treated with 3 doses of pulse steroid (500 mg) and tacrolimus was switched to sirolimus. Serum creatinine decreased to 1.3 mg/dl but later had gradual rise of serum creatinine over 2 years from 1.3 to 2.3 mg/dl till last month with subnephrotic proteinuria of 500 mg/day. Kidney biopsy was done in January 2019 which showed diffuse infiltration of interstitium and vascular area with pale eosinophilic material strongly positive for Congo red stain suggestive of amyloid [Figures 1 and 2] and changes of chronic calcineurin inhibitor toxicity. Serum amyloid-associated protein (SAA) and immunofluorescence study was negative for immunoglobulins and complements. Immunohistochemistry with anti LECT2 antibodies showed diffuse positivity within amyloid deposits [Figure 3]. Electron microscopy was attempted on the paraffin block but due to extensive processing artifacts, it was not interpretable. Serum protein electrophoresis was negative for monoclonal proteins. Serologies including hepatitis panel, antinuclear and antineutrophilic cytoplasmic antibodies, and complements were within normal range. The first biopsy was reevaluated and showed very minimal PAS negative material in the interstitium, but due to insufficient tissue, Congo red could not be performed for confirmation. His father (donor) was also evaluated to rule out any donor source of the deposits, but he was clinically asymptomatic with normal renal function and no proteinuria. The patient had no hepatomegaly on ultrasound and his liver function tests were normal, thus ruling out liver involvement. He was managed conservatively with angiotensin converting enzyme inhibitor and immunosuppression continued. Serum creatinine on the last follow-up was stable at 2.5 mg/dl.

- Extensive deposits of weak PAS positive material along tubular basement membrane and interstitium characteristic of amyloid (magnification 40×)

- Congo Red Staining of the deposited material along tubular basement membrane and in the interstitium
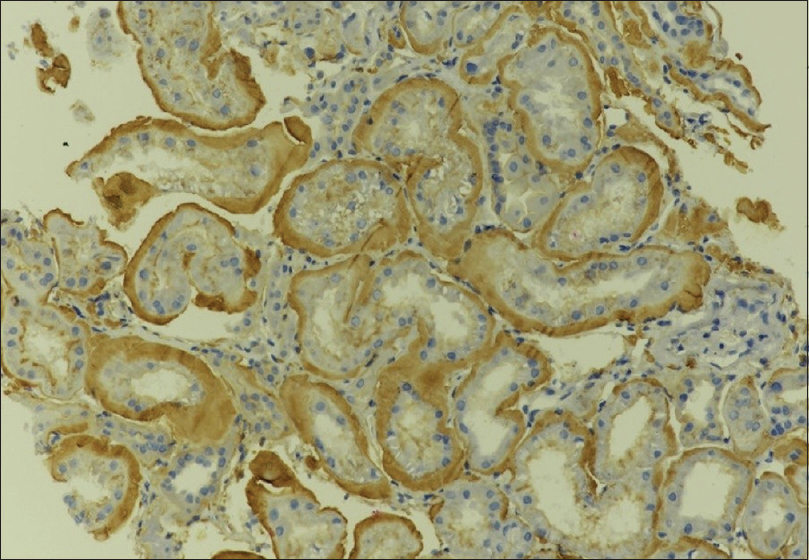
- Strong staining for LECT2 by immunohistochemistry along tubular basement membrane and interstitial deposits
Discussion
LECT2 Amyloidosis is the fourth most common type of amyloidosis with a prevalence of 3.5% in the largest amyloid cohort of 4139 cases described by Mayo Clinic.[5] The LECT2 gene has been mapped to chromosome 5q31. Patients with homozygous G allele with single amino acid switch at position 40 are predisposed to develop LECT2 amyloidosis.[6] Most patients are elderly and present with mild-to-moderate renal insufficiency and variable proteinuria. In contrast to AL and AA amyloidosis, LECT2 amyloidosis is a relatively benign disease associated with a slow GFR decline and minimal proteinuria.[34] Hepatic ALECT2 amyloidosis most commonly presents with elevation of alkaline phosphatase.[7] Histologically, it is typically deposited in interstitium and mesangial regions with strong congophilia.[38] This is in contrast to AL and AA amyloidosis in which glomerular and vessels are the major site of amyloid deposition. Contrary to Apo AI amyloidosis and Apo AIV amyloidosis, which mainly affect the medullary interstitium, ALECT2 amyloidosis shows a predominant involvement of cortical interstitium.[34] Immunofluorescence is usually negative. A minority (9%) of patients with ALECT2 amyloidosis show false-positive staining for serum amyloid A (SAA).[4] Amyloid typing by immunohistochemistry using commercially available antibodies against LECT2 is a highly sensitive diagnostic tool.[9] Typing the amyloid deposits by mass spectroscopy-based proteomics is, however, the best tool to diagnose LECT2 amyloidosis, which is available in limited centers only.[10] Patient survival is superior to that seen in renal AL and AA, presumably because of the absence of cardiac involvement. Currently no effective treatment is available for this disease.[6]
Conclusion
To conclude, we should suspect this entity when the deposition of amyloid material is more in interstitium and vessel walls in comparison to glomerular component with prominent congophilia.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Leukocyte chemotactic factor 2: A novel renal amyloid protein. Kidney Int. 2008;74:218-22.
- [Google Scholar]
- Clinical, morphologic, and genetic features of renal leukocyte chemotactic factor 2 amyloidosis. Kidney Int. 2014;86:378-82.
- [Google Scholar]
- Characterization and outcomes of renal leukocyte chemotactic factor 2-associated amyloidosis. Kidney Int. 2014;86:370-7.
- [Google Scholar]
- Proteome of amyloidosis: Mayo Clinic experience in 4139 cases. Blood. 2013;122:1900.
- [Google Scholar]
- Leukocyte cell-derived chemotaxin 2-associated amyloidosis: A recently recognized disease with distinct clinicopathologic characteristics. Clin J Am Soc Nephrol. 2015;10:2084-93.
- [Google Scholar]
- Leukocyte cell-derived chemotaxin 2 (LECT2)-associated amyloidosis is a frequent cause of hepatic amyloidosis in the United States. Blood. 2014;123:1479-82.
- [Google Scholar]
- Prevalence and morphology of leukocyte chemotactic factor 2-associated amyloid in renal biopsies. Kidney Int. 2010;77:816-9.
- [Google Scholar]
- Leukocyte chemotactic factor 2 amyloidosis can be reliably diagnosed by immunohistochemical staining. Hum Pathol. 2014;45:2179.
- [Google Scholar]
- Laser microdissection and mass spectrometry-based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int. 2012;82:226-34.
- [Google Scholar]




