

Translate this page into:
IgA Nephropathy: Emerging Mechanisms of Disease
Corresponding author: Haresh Selvaskandan, John Walls Renal Unit, University Hospitals Leicester NHS Trust, Leicester, United Kingdom. E-mail: hs328@leicester.ac.uk
-
Received: ,
Accepted: ,
How to cite this article: Roberts LE, Williams CEC, Oni L, Barratt J, Selvaskandan H. IgA Nephropathy: Emerging Mechanisms of Disease. Indian J Nephrol. 2024;34:297-309. doi: 10.25259/ijn_425_23
Abstract
Immunoglobulin A nephropathy (IgAN) is the most common primary glomerulonephritis reported across the world and is characterized by immunoglobulin A (IgA) dominant mesangial deposits, which are poorly O-glycosylated. This deposition leads to a cascade of glomerular and tubulointerstitial inflammation and fibrosis, which can progress to chronic kidney disease. The variability in rate of progression reflects the many genetic and environmental factors that drive IgAN. Here, we summarize the contemporary understanding of the disease mechanisms that drive IgAN and provide an overview of new and emerging therapies, which target these mechanisms.
Keywords
Four-hit hypothesis
IgA nephropathy
inflammation
pathogenesis
treatments

Introduction
Immunoglobulin A nephropathy (IgAN) is a common cause of primary glomerulonephritis.1-3 It is characterized by IgA-dominant mesangial deposits, poorly O-glycosylated at their hinge region, which drive a variable inflammatory and fibrotic response within the kidney.4 This variability manifests as a spectrum of clinical presentations, ranging from isolated non-visible hematuria to rapidly progressive glomerulonephritis.4-6 In the largest reported national registry of IgAN, approximately 50% of children and 75% of adults with IgAN progressed to kidney failure (KF) within 20 years of diagnosis, with progression occurring even in those with low levels of proteinuria (<1g/day) who were previously considered low risk.7 The incidence and severity of IgAN also vary with ethnicity, being more frequent among Pacific Asians compared with Caucasians, and being particularly rare among those of African ancestry.8-10
The heterogeneity of IgAN reflects the variety of mechanisms that drive it, involving an interplay between genetic and environmental factors, including genetic risk loci, epigenetic controllers, the microbiome, the complement system, mucosally primed B cells, and local inflammatory and fibrotic signaling pathways acting in concert.11-14 Here, we review contemporary insights into IgAN pathophysiology and provide a brief overview of treatments leveraging these insights for therapeutic benefit.
Immunoglobulin A — An Overview
IgA is the dominant antibody of mucosal surfaces and exists as two isoforms in humans, IgA1 and IgA2 [Figure 1].15 While IgA1 is dominant in the serum, the ratio of the two isoforms varies at mucosal surfaces, with proportions equalizing in the distal ileum.16 The isoforms differ at the hinge region, between the first and second constant domains of the α1 heavy chain. The IgA1 hinge is longer by 13 amino acids, nine of which are serine and threonine residues, which have the potential to be O-glycosylated.17 This elongation may confer enhanced antigen recognition but also renders IgA1 susceptible to proteolytic cleavage by proteases produced by bacteria, including Streptococcus pneumoniae and Haemophilus influenzae,18 a feature that has been exploited for therapeutic benefit in some animal models.19
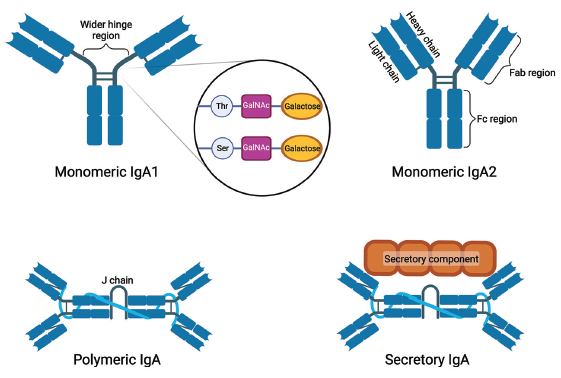
- States of human IgA. Monomeric IgA is formed from two heavy chains and two light chains, with a hinge region separating the antigen binding (Fab) and effector domains (Fc). The hinge region of monomeric IgA1 is wider than that of IgA2, with additional serine and threonine residues that can undergo O-linked glycosylation. Polymeric IgA (pIgA) mainly exists in the form of dimers of IgA that are covalently joined by a peptide called “J chain”. Secretory IgA is formed following the trafficking of pIgA to mucosal surfaces, with the extracellular portion of the polymeric Ig receptor remaining bound to the pIgA as the “secretory component”. Thr: threonine; Ser: serine; GalNAc: N-acetylgalactosamine. Figure created with BioRender.com.
IgA1 can exist in monomeric or polymeric forms Figure 1, adapted from Selvaskakandan et al.14. Monomeric IgA1 predominates in serum, while polymeric IgA1 (pIgA; large dimers of IgA covalently joined by a joining chain, or “J chain”) is found in mucosal secretions.20 pIgA is trafficked to mucosal surfaces by the polymeric Ig receptor, the extracellular portion of which (referred to as the “secretory component”) remains coupled to pIgA after trafficking forming secretory IgA (sIgA). The secretory component is thought to stabilize pIgA by enhancing its resistance to proteolysis.20
The Four-Hit Hypothesis
A hallmark of IgAN is a disturbance in the production of O-glycosylated IgA1. This is the initiating step of a sequence of events thought to be necessary for disease to develop. These events are framed by the four-hit hypothesis and include the following:5
The appearance of poorly O-glycosylated IgA1 (also referred to as galactose-deficient IgA1 or Gd-IgA1).
The formation of IgG or IgA autoantibodies against Gd-IgA1.
The formation of Gd-IgA1-containing immune complexes (Gd-IgA1 CICs).
An accumulation of Gd-IgA1 CICs in the glomerular mesangium, leading to inflammation and fibrosis.
Gd-IgA1
The O-glycosylation of IgA1 begins with the addition of N-acetylgalactosamine (GalNAc) to threonine and/or serine residues by N-acetylgalactosaminyltransferase 2 (GALNT2). GalNAc is then O-galactosylated by core-1-ß1,3-galactosyltransferase (C1GALT1) supported by the chaperone foldase Cosmc.21 This is disrupted in IgAN, leading to an elevated proportion of Gd-IgA1 in the serum.5,22,23 The tendency to produce Gd-IgA1 has a high degree of heritability — 40–50% of first-degree relatives of those with IgAN have elevated levels of circulating Gd-IgA1,24,25 and quantitative trait (QT) genome-wide association (GWA) studies have demonstrated that a haplotype spanning the C1GALT1 gene is strongly associated with elevated Gd-IgA1 levels.26 Environmental factors also seem capable of triggering C1GALT1 and Cosmc dysregulation:14 IgD, the only other O-glycosylated immunoglobulin in humans, remains normally galactosylated in IgAN, indicating that the dysregulation appears after IgA class switch recombination takes place.27 The appearance of Gd-IgA1 is considered hit one and on its own does not seem sufficient to produce clinically significant disease.28
Anti-Gd-IgA1
The presence of IgG and IgA anti-Gd-IgA1 antibodies may be explained by two separate but complementary processes. A somatic mutation in the gene-encoding IgG (alanine-to-serine substitution within the complementarity determining 3 (CD3) domain of the variable region) has been shown to result in IgG with a specificity for the hinge region of Gd-IgA1.29 Firstly, the exposed GalNAc residues of Gd-IgA1 may act as an autoantigen, triggering an IgA or IgG autoantibody response in those susceptible.30 Secondly, IgG and IgA antibodies generated as a natural response to GalNAc-containing glycoproteins on the surface of pathogens may also function as anti-Gd-IgA1 antibodies by cross-reacting with the Gd-IgA1 hinge region.31 Circulating anti-Gd-IgA1 levels are raised in IgAN and correlate with disease progression,30 and when present, co-deposition of IgG in the mesangium is associated with a poorer prognosis.32 However, the variable presence of mesangial IgG in IgAN may suggest that IgG anti-Gd-IgA1 is not an absolute requirement for disease.
Gd-IgA1 CICs
Gd-IgA1 CICs are elevated in the serum in IgAN and correlate with the extent of hematuria, proteinuria, and estimated glomerular filtration rate (eGFR).33,34 Gd-IgA1 CICs can form when anti-Gd-IgA1 antibodies bind Gd-IgA133 or when Gd-IgA1 forms non-covalent self-aggregates (the usual glycosylation of IgA1 protects against self-aggregation).35 Immune complexes may also form as Gd-IgA1-soluble CD89 complexes.36 CD89 is expressed on the cell surface of myeloid cells and functions as an Fc receptor for IgA. IgA binding can trigger CD89 shedding, with subsequent Gd-IgA1-soluble CD89 immune complex formation.36,37 Although no CD89 homolog is found in mice, a transgenic mouse model expressing human CD89 spontaneously developed an IgAN-like phenotype, indicating a possible role in disease development.38
Mesangial deposition of IgA1
IgA1 deposition likely occurs due to specific interactions between the epitopes in Gd-IgA1 CICs and glomerular basement membrane and mesangial matrix proteins, although the precise proteins involved are poorly understood. Once deposited, these complexes bind to mesangial cell surface receptors, including the transferrin receptor (CD71), soluble CD89, transglutaminase-2 (TGase2), and β-1,4-galactosyltransferase-1, all of which can be upregulated in IgAN.39,40
Mouse models and in vitro work demonstrate that IgA1-soluble CD89 complexes interact with mesangial CD71. This leads to further CD71 expression via the calcium-dependent enzyme TGase2, further enhancing IgA1 deposition.39,41
β-1,4-galactosyltransferase-1 is constitutively expressed on mesangial cells and binds the Fc portion of IgA1. Mesangial cells incubated with anti-β-1,4-galactosyltransferase antibodies bind less IgA and display a blunted inflammatory interleukin (IL)-6 response and reduced phosphorylation of spleen tyrosine kinase (Syk),40 a non-receptor tyrosine kinase, which coordinates mesangial cell proliferation and cytokine secretion.42
Exposed mesangial βII-spectrin directly binds IgA in the ddY spontaneous IgAN mouse model. Anti-βII-spectrin IgA was found in the sera of up to 60% of a cohort of Japanese patients with IgAN (n = 45), suggesting that the development of autoantibodies to mesangial antigens may contribute to disease pathogenesis.43 Although this suggests that immune complex formation in sera may not be essential for IgAN to develop, in vitro evidence indicates that circulating immune complexes are likely to play a role in most patients with IgAN. IgA1 CICs induce mesangial proliferation and inflammation while monomeric IgA1 does not.13,31 However, it must be noted that IgA1 CIC mesangial deposits can also be observed in individuals without clinically apparent disease,44 suggesting that the right combination of Gd-IgA1 CICs and a predisposing local glomerular environment may be needed for the disease to develop. In those that do develop disease, a spectrum of histopathological lesions are observed.45 Reflecting this, there is significant heterogeneity in the degree of inflammation and fibrosis in IgAN, which can occur through several pathways.4,6
Mediators of Local Inflammation and Fibrosis
Cytokines, chemokines, and inflammatory pathways in IgAN
Inflammation and fibrosis in IgAN can be driven by pro-inflammatory cytokines, proteinuric tubular damage, and the complement system, which act together to drive disease.
Mesangial Gd-IgA1 CIC deposition triggers mesangial proliferation, mesangial matrix expansion, and interstitial macrophage infiltration.38,46 Deposition also induces the mesangial production of pro-inflammatory or fibrotic cytokines (including tumor necrosis factor (TNF)-α, IL-6, and transforming growth factor (TGF)-β) and activates nuclear transcription factor kappa B (NF-κB), which can promote an influx of pro-inflammatory cells.47,48 TNF-α production acts as an autocrine stimulus, promoting podocyte production of TNF-α and IL-6, and in turn generates an inflammatory positive feedback loop.48 The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/ mechanistic target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK) 1/2 kinase pathways are key signaling pathways and have been shown to be activated in mesangial cells following exposure to pIgA, an effect likely mediated through binding to CD71. Activation leads to mesangial proliferation and cytokine production.49 Syk is also implicated in IgAN and can be activated by IgA binding to mesangial β-1,4-galactosyltransferase-1. Syk activation results in mesangial cell proliferation and production of IL-650 and is upregulated in glomeruli in IgAN, correlating with serum creatinine.40,51
The magnitude of the inflammatory response to IgA CICs may also relate to their physical properties, which may partly account for the variation in severity of inflammation or fibrosis observed. Larger (>800 kDa) Gd-IgA1 CICs promote greater mesangial cell activation, possibly due to greater cell surface receptor cross-linking, leading to enhanced IL-6 and IL-8 secretion and an over-production of the extracellular matrix protein laminin.52
The pro-inflammatory cytokine milieu triggered by mesangial activation can also facilitate mesangial–podocyte cross-talk. Mesangial cell-derived chemokine C-X-C motif ligand-1 (CXCL1), TNF-α, and TGF-β1 suppress nephrin and erzin expression in cultured podocytes, directly impacting slit diaphragm integrity.47,53 TNF-α and TGF-β1 also promote podocyte injury, leading to podocyte death in vitro.47
Angiotensin II is also released by mesangial cells in response to IgA1 deposition, and this can reduce podocyte α3β1 integrin expression, impairing podocyte adhesion.54 Podocyte injury ultimately leads to segmental glomerulosclerosis, which compounds glomerular injury due to mesangial inflammation and accelerates the loss of kidney function.48,55 The collective response to mesangial IgA accumulation is an increase in glomerular permeability and the development of worsening proteinuria, which in itself promotes downstream tubular damage.48,56
Tubular injury
Albumin and Gd-IgA1 CICs cross the damaged glomerular filtration barrier and stimulate proximal tubular epithelial cells to produce cytokines and growth factors, which induce tubulointerstitial inflammation and macrophage infiltration.57 Further tubular damage can be mediated by mesangial-derived TNF-α.58 Infiltrating macrophages drive tubulointerstitial fibrosis through the release of profibrotic cytokines and inhibitors of matrix-degrading proteases, such as tissue inhibitor of metalloproteinase-1 and plasminogen activator inhibitor,59 and through direct interactions with collecting duct epithelial cells.60
The complement system
The complement system is a set of soluble and membrane-associated proteins, which rapidly activate in an amplifying cascade to augment local immune responses by contributing to pathogen opsonization and inflammation [Figure 2, adapted from Girardi et al.].61,62 Complement activation occurs via three pathways:
-
1.
The alternative pathway (AP) — via spontaneous hydrolysis and moieties on microbial surfaces.
-
2.
The lectin pathway (LP) — via the recognition of mannose and fucose residues on microbial surfaces.
-
3.
The classical pathway (CP) — via antigen–antibody complexes.
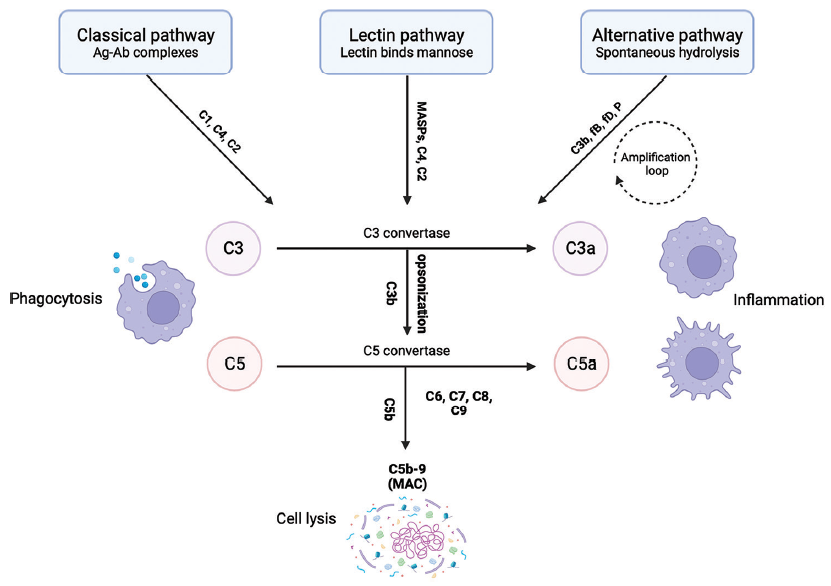
- Schematic, simplified model of the complement pathway. MASPs: MBL-associated serine proteases; CFD: complement factor D; CFB: complement factor B; P: properdin; MAC: membrane attack complex. Figure created with BioRender.com
A number of studies have shown that the extent of complement activation, measured as glomerular C3 deposition, correlates with disease severity and progression.63 The absence of C1q deposition64 suggests that the activation of the complement cascade does not involve the CP in IgAN.65,66
AP activation is supported by the frequent finding of mesangial C3, along with AP regulators, including properdin and factor H-related protein 5 (FHR5).67,68 Genetic variants recognized to result in impaired AP regulation, in particular in the factor H: complement factor H-related proteins axis, are associated with IgAN.69 Although the precise mechanisms by which the AP is activated in IgAN remain incompletely understood,70,71 it is clear that the AP is a driver of mesangial inflammation.
LP activation in IgAN is indicated by mesangial C3 and C4d deposition in the absence of C1q alongside deposition of mannose-binding lectin (MBL) and MBL-associated serine protease 2 (MASP-2).63,72 The exposed GalNAc residues of polymeric Gd-IgA1 may trigger LP activation. The LP is activated in vitro by pIgA purified from patients with IgAN, but not mIgA, and is inhibited when pIgA is incubated with saccharides.73 LP activation in IgAN results in glomerular inflammation — extent of C4d deposition correlates with the severity of glomerular injury, degree of proteinuria, and risk of progression to KF in IgAN.63,72
Several clinical trials are currently underway exploring the efficacy of complement-modulating therapies in IgAN and are summarized further in this review (see Modulating the complement system).
Mucosal Immunity and IgAN
Visible hematuria can occur following mucosal infections in IgAN, indicating a link between the mucosal immune system and nephritis in IgAN. IgA is produced primarily as pIgA at mucosal surfaces, arising from lamina propria-sited plasma cells of the mucosa-associated lymphoid tissue (MALT).74,75 Serum pIgA and sIgA (exclusively produced at mucosal surfaces) are elevated in IgAN, are found in glomeruli in IgAN, and are associated with disease severity,76,77 supporting an important role of the MALT in IgAN.
The precise mechanisms by which “mucosal” IgA enters the serum are unclear. Naïve mucosal B cells are activated locally through T-cell-dependent and T-cell-independent mechanisms78 and then enter the lymphatics and home to systemic sites where they undergo maturation. Mature B cells then re-home back to the mucosa where they remain as IgA-producing plasma cells. One hypothesis is that, in IgAN, these maturing B cells mis-home to the bone marrow and release “mucosal” polymeric Gd-IgA1 into the circulation.74 Alternatively, pIgA/sIgA produced by MALT-residing plasma cells may be “reverse trafficked” from mucosal surfaces back into the serum.74,79 IgA-producing plasmablasts and plasma cells, which are CD20-negative, have been found in the lamina propria of the gut. If these cells are the major source of Gd-IgA1 in IgAN, it may account for the lack of efficacy of rituximab (a monoclonal antibody (mAb) against CD20) in IgAN, despite its benefits in a variety of other antibody-mediated glomerular diseases.80,81
Microbiome Involvement in IgAN
The gut microbiome is altered in IgAN patients compared with healthy subjects, with relative depletion of host-beneficial bacteria.82,83 Reflecting this, the fecal microbiomes of Chinese adults with IgAN were different compared to healthy subjects — those with IgAN had higher levels of Bacteroides and Escherichia–Shigella and lower levels of Bifidobacterium and Blautia spp. These patterns are associated with hematuria and proteinuria severity.83 Other studies have also reported intestinal, tonsillar, and oral microbiome disturbances in IgAN.84-88
Patients with IgAN concomitantly infected with Helicobacter pylori produce an exaggerated polymeric Gd-IgA1 response compared to healthy subjects. When compared to systemic antigen exposure, both groups produced a normal equal IgA1 response, indicating that immune dysregulation in IgAN is limited to the MALT.89,90
Clear mechanistic links between microbiome dysbiosis and IgAN in humans are limited; however, there is evidence from animal models. B-cell-activating factor (BAFF) transgenic mice (which overexpress BAFF, a key modulator of B-cell activation) develop a kidney disease identical to IgAN in association with elevated serum pIgA and an increase in IgA+ plasma cells in the gut lamina propria. When raised in germ-free conditions, the mice fail to develop a gut microbiome, the serum IgA levels are significantly reduced, and there are no mesangial IgA deposits.91 Transferring these mice back into the normal environment allows the development of a gut microbiome and restores the IgAN-like phenotype.
B-cell activation in the MALT can occur through T-cell-dependent and T-cell-independent pathways. T-cell-independent B-cell activation involves the cytokines BAFF and a proliferation-inducing ligand (APRIL), which are both members of the TNF superfamily [Figure 3, adapted from Kaegi et al].92-94 Both can be produced by innate immune cells in response to the activation of toll-like receptors (TLRs) by pathogen-associated molecular patterns (PAMPs) released by the gut microbiome, and both play key roles in driving mucosal B-cell maturation and IgA class switch recombination. BAFF and APRIL act via three cell surface receptors: the BAFF receptor (BAFF-R), B-cell maturation antigen (BCMA), and transmembrane activator and CAML interactor (TACI).93
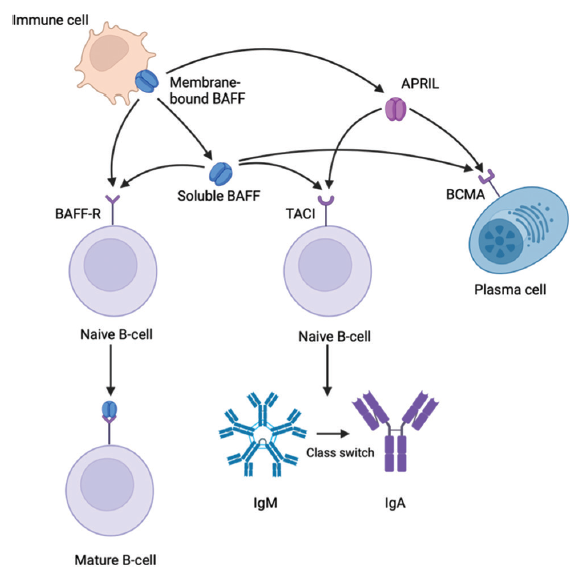
- Schematic representation of the role of B-cell-activating factor (BAFF), a proliferation-inducing ligand (APRIL), and their receptors BAFF receptor (BAFF-R), transmembrane activator and CAML interactor (TACI), and B-cell maturation antigen (BCMA). These receptors are expressed on B cells and plasma cells, and APRIL can only bind to TACI and BCMA, while BAFF can bind to all three receptors. The binding of BAFF to BAFF-R promotes the survival and maturation of naïve B cells. TACI allows IgA class switching of immunoglobulin M (IgM) to immunoglobulin G (IgG) and T-cell independent B-cell responses to antigens. BCMA mediates plasma cell homeostasis. Figure created with BioRender.com
Toll-like receptors
Mucosal B cells express a variety of TLRs, including TLR4, TLR9, and TLR10, which have each been implicated in IgAN.95-97 Expression of TLR4 in circulating peripheral blood mononuclear cells (PBMCs) in children with IgAN is increased compared with healthy subjects,97 and TLR10 is upregulated in Korean children with IgAN who carry the single-nucleotide polymorphism (SNP) rs10004195. Increased TLR10 expression is associated with more proteinuria.96 The ddY mouse has elevated levels of serum TLR9, which associates with a more severe IgA-mediated kidney disease.95 TLR activation has also been shown to reduce C1GALT1 activity by promoting methylation of the Cosmc gene. Furthermore, the activation of TLR4 in peripheral B cells promotes the production of Gd-IgA1.98,99 These data suggest that TLRs form part of the mechanistic link between the mucosal microbiome and B-cell activation in IgAN.
BAFF and APRIL
Serum BAFF and APRIL are elevated in IgAN, correlating with worse histology and poorer kidney function.100 TNFSF13, which encodes APRIL, has been identified as a susceptibility locus in a GWA study in adult IgAN, which also found a correlation between this variant and higher serum IgA concentrations.101 Similarly, increased levels of APRIL may be associated with higher serum levels of Gd-IgA1 and a more severe phenotype of IgAN.98 The sources of serum BAFF and APRIL in IgAN are not known, but it is tempting to speculate that serum levels may reflect the degree of activity within the MALT, and this needs to be confirmed.
Taken together, these findings support a role for the mucosal microbiome in IgAN.
Genetic Determinants of IgAN
Epidemiological observations suggest a genetic contribution to both the development of IgAN and the determination of the severity of disease. IgAN incidence is higher in Pacific Asia compared with Western Europe and North America and is relatively rare in sub-Saharan Africa.9,10,102 Risk of KF among Pacific Asians with IgAN living in North America also appears to be higher than that of other ancestries living in the same regions.103
A large GWA study highlighted a panel of IgAN risk loci, many of which support the contemporary understanding of IgAN pathophysiology. Loci included TNFSF13, which encodes APRIL, integrin subunit alpha M (ITGAM), and integrin subunit alpha X (ITGAX), which encode dendritic cell integrins, which can modulate gut plasma cell IgA production in mouse models, and a panel of other loci, including CARD9, which modulates gut mucosal immunity and integrity.104 The frequency of these loci varies geographically, associating with local microbial diversity. A more recent GWA study that included over 10,000 biopsy-confirmed IgAN cases and over 28,000 controls identified 30 risk loci, including 16 not previously reported.105 These loci included 14 alleles that have roles in pathways that could be targeted by therapies currently in development, including CFH, which encodes complement factor H, RELA, which encodes nuclear kappa B p65, and TNFSF13 (APRIL) and TNFRSF13B, which encodes TACI.105 The cumulative frequency of risk loci predicted an earlier onset of KF.105
Epigenetic Mediators of IgAN: Micro-RNAs (miRs)
miRs are short noncoding oligonucleotides, which suppress gene expression by hybridizing with target mRNAs, thereby preventing translation (either at initiation or at elongation stages), or triggering mRNA degradation [Figure 4].106 Since their relatively recent discovery in 1993, miRs have been implicated in the pathophysiology of several chronic diseases, including kidney disease.107-109
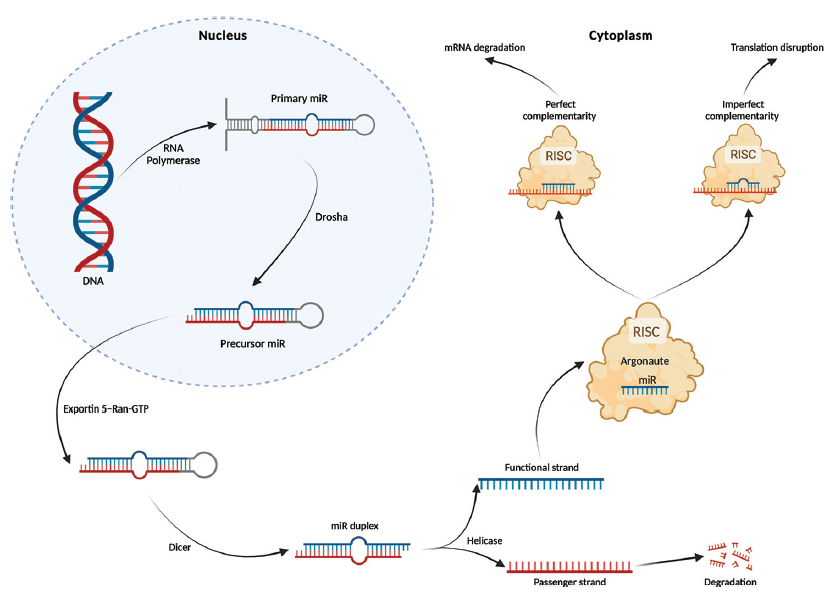
- Overview of miR biogenesis. The process of miR formation is initiated by RNA polymerase, which generates a primary miR transcript from nuclear DNA. The nuclease Drosha then cleaves the 5´ and 3´ regions of the initial hairpin transcript to form precursor miR (pre-miR), which is transported out of the nucleus coupled with an exportin 5-ran-GTP complex. Once in the cytoplasm, RNase Dicer cleaves the loop region of the pre-miR hairpin to form double-stranded RNA, which subsequently is separated by a helicase to form a passenger strand that undergoes degradation and a functional guide strand. The functional strand becomes incorporated in the RNA-induced silencing complex (RISC) and acts to guide the RISC to target mRNA. The seed region of the functional strand consists of a sequence of 6 to 8 nucleotides usually starting at position 2, and it is this that determines the mechanism by which RISC silences gene expression. Translation is disrupted if the seed region imperfectly hybridizes with the mRNA target, whereas complete complementarity causes the argonaute protein of the RISC to trigger mRNA degradation. A single miR is thus capable of modulating the expression of many genes to alter cellular physiology. Figure created with BioRender.com
miR expression is dysregulated in blood, urine, and kidney tissue in IgAN, and although several miRs have been proposed as biomarkers for IgAN, only a few have been validated in independent cohorts.110
In two small, but independent, cohorts of Italian, Greek, Chinese, and Japanese patients, serum levels of miRs let-7b and miR-148b were elevated in IgAN and were able to distinguish IgAN from healthy and disease controls (minimal change disease, focal segmental glomerulosclerosis, and membranoproliferative glomerulonephritis).111 A subsequent study of the Greek participants from the initial validation cohort found that serum let-7b, as well as models combining let-7b and 148b at the time of diagnosis, could predict kidney function decline at the end of follow-up (mean 11.9 years).112 miRs 148b and let-7b are predicted to suppress the translation of GALNT2 and C1GALT1, respectively, both enzymes that are instrumental in IgA1 hinge region O-glycosylation.113,114
miR-21 was raised in the urinary sediment of Chinese patients with IgAN compared with healthy subjects;115 however, urinary miR-21 levels were not able to distinguish between IgAN and hypertensive nephropathy in a separate Chinese cohort.116 These findings are consistent with other work implicating miR-21 in the final common pathway of kidney fibrosis.117
Intrarenal miR expression has also been investigated using kidney biopsy specimens. Kidney expression of five miRs (150-5p, 155-5p, 146b-5p, 135a-5p, and 204) was dysregulated in IgAN, and when added, two of these miRs improved the prognostic prediction accuracy of the International IgAN Risk Prediction Tool (IIGANRPT), the current gold standard for predicting risk of progression in IgAN.118-120 Like miR-21, dysregulation of these miRs has been described across a range of kidney diseases, indicating that they likely regulate final common pathways of inflammation and fibrosis.121-125
Overview of Treatments/Trials Leveraging Novel Insights into Pathophysiology
Following collaborative work led by the Kidney Health Initiative, which demonstrated a strong association between an early treatment effect on proteinuria and a composite of traditional endpoints (doubling of serum creatinine, KF, and death), an early change in proteinuria has been accepted as a reasonable surrogate endpoint for progression to KF in IgAN.126 Drugs can now be considered for accelerated approval based on the observed proteinuria change between 6 and 12 months. Confirmation of longer-term kidney function protection is required in all studies and is being evaluated as the change in eGFR over a 2-year follow-up period. This change in the regulatory environment has resulted in an explosion in clinical trial activity in IgAN. Coupled with evolving pathophysiological insights, this has led to a welcomed upsurge in therapeutics undergoing evaluation for IgAN (detailed review127).
Modulating mucosal immunity: TRF-budesonide (Nefecon)
Nefecon is a corticosteroid packed in a starch capsule, which releases its contents at the terminal ileum, an area with a high concentration of Peyer’s patches.128 Nefecon suppresses serum BAFF and APRIL in a dose-dependent fashion, reduces circulating Gd-IgA1, sIgA, and IgA–IgG complexes, and modifies the composition of IgA-containing immune complexes in IgAN.129,130 Nefecon produced a 27.3% reduction in mean urine protein-to-creatinine ratio (uPCR) by 9 months in the phase 2 NEFIGAN trial and preserved kidney function at 12 months.131 The results were supported by the phase 3 NefIgArd trial, where, in part A of the study, 16 mg once a day of Nefecon produced a 27% reduction in mean uPCR and conferred a protective effect on kidney function (3.87 mL/min/1.72 m2) at 9 months.132 Benefits persisted after treatment at 24 months, although a rise in proteinuria was noted on cessation of Nefecon after 12 months.133 Nefecon undergoes rapid first-pass metabolism, minimizing the systemic absorption of budesonide, and was generally well tolerated in both the NEFIGAN and NefIgArd trials. In 2022, Nefecon became the first drug to receive approval from the Food and Drug Administration (FDA) and European Medicine Agency (EMA) for the treatment of IgAN at high risk of progression.
Modulating the mucosal microbiome
Fecal microbiota transplantation (FMT) has been investigated as a treatment for conditions associated with gut dysbiosis, including recurrent or persistent Clostridium difficile infections, with the aim of re-establishing a balance in gut flora.134 Two patients with refractory IgAN (proteinuria of 880 mg/day and 2020mg/day despite combinations of glucocorticoids, tacrolimus, and mycophenolate mofetil) were treated with a FMT.135 The first patient received fresh FMT 40 times consecutively (200 mL daily, 5 days a week for 8 weeks), followed by a further 57 times (200 mL daily, 10–15 days per month) over 5 months, and achieved a 37.0% reduction in 24-hour urinary protein three months post-treatment. The second patient received 60 fresh FMTs (200 mL daily, 10–15 days per month) over 6 months achieving a reduction of 62.8% in 24-hour urinary protein, which remained stable for 6 months post-treatment.
Two small, single-arm trials (n = 30, NCT03633864, and n = 15, NCT05182775) are evaluating FMT in adults with biopsy-proven IgAN who are not responsive to or unable to tolerate glucocorticoids or immunosuppressants.
Modulating the complement system
Therapies currently being evaluated in IgAN target the AP, the LP, and the common terminal pathway.
Inhibitors of the AP are directed against factors B and D
Factor B. Iptacopan, a small molecule oral factor B inhibitor, has been evaluated in a phase 2 clinical trial, which demonstrated a 23% reduction in proteinuria at three months compared with placebo. Iptacopan is now being assessed in a phase 3 randomized controlled trial (APPLAUSE-IgAN, NCT04578834).136,137 IONIS-FB-LRx is an antisense oligonucleotide, which suppresses hepatic factor B production. In a small open-label phase 2 study, IONIS-FB-LRx suppressed AP activity and produced a 44% reduction in proteinuria by week 29. IONIS-FB-LRx will be evaluated in a phase 3 trial soon (IMAGINATION; NCT05797610). Factor D. The small molecule oral factor D inhibitor, vemircopan, is currently being assessed in a phase 2 trial (NCT05097989).
Inhibition of the LP is directed to blocking MASP-2 activity
Narsoplimab is a mAb directed against MASP-2 preventing the formation of the C2bC4b C3 convertase. Narsoplimab was evaluated in a small phase 2 trial involving four patients with IgAN concurrently receiving corticosteroids (sub-study 1) and twelve patients with IgAN not receiving corticosteroids (sub-study 2). Sub-study 1 was a single-arm trial in which narsoplimab produced a 72% decline in mean proteinuria when added to RAS blockade (corticosteroids were tapered on enrolment). Sub-study 2 was a placebo-controlled randomised controlled trial (RCT), and although there was no difference in proteinuria reduction between the groups, twelve patients from both studies who continued to a dosing extension phase (median follow-up 22 months) experienced a 38% reduction in proteinuria from baseline and a reduced rate of eGFR decline (decline of 5.2 (±2.1) mL/min/yr vs 8.6 (±3.7) mL/min/yr in placebo).138
Drugs targeting the final common pathway inhibit C3 or C5 activity
C3 inhibitors include pegcetacoplan and ARO-C3. Pegcetacoplan (APL-2) is a peptide inhibitor of both C3 and C3b. It is being evaluated in a phase 2 basket trial assessing efficacy in various glomerulopathies, including in IgAN (NCT03453619). ARO-C3 is an antisense oligonucleotide, which suppresses hepatic factor C3 production. It is currently being evaluated in a global phase 1/2 study (NCT05083364).
C5 inhibitors include ravulizumab and cemdisiran. Ravulizumab is a recombinant mAb directed against C5 being evaluated in an ongoing phase 2 double-blinded RCT, which includes patients with IgAN and lupus nephritis (NCT04564339). Cemdisiran (ALN-CC5) is an RNA interference (RNAi) therapy, which suppresses hepatic C5 production. It is conjugated to GalNAc, allowing it to bind hepatic asialoglycoprotein receptors, meaning it can be targeted to the liver. It was evaluated in a small phase 2 RCT for IgAN (n = 31), where it produced a 37% mean reduction in proteinuria from baseline compared with placebo at 32 weeks (NCT03841448).
C5a receptor antagonism has also been evaluated in IgAN
Avacopan is a selective C5a receptor antagonist, which was evaluated in a small trial of seven patients with IgAN. It produced an approximately 50% decline in uPCR from baseline in three of the seven enrolled after 12 weeks of treatment, reduced urinary markers of inflammation (monocyte chemoattractant protein 1), and was well tolerated.139
Therapies directed against B cells
Therapies that regulate B-cell activity can be broadly considered to be B-cell depleting or B-cell modulating.
B-cell-depleting therapies include rituximab (CD20), felzartamab (CD38), and mezagitamab (CD38). Rituximab failed to demonstrate efficacy in IgAN in a small RCT.81 Felzartamab (MOR202) is being evaluated in a double-blinded phase 2 RCT (n = 54) (NCT05065970), and mezagitamab (TAK-079, another mAb against CD38), is being evaluated in a small phase 1b IgAN study (NCT05174221). The efficacy of these monoclonal antibodies would support a role for CD38+ plasma cells in the production of Gd-IgA1 in IgAN.140
B-cell-modulating therapies are directed against BAFF, APRIL, or both. BAFF inhibitors include blisibimod and belimumab. Blisibimod is a peptibody inhibitor of BAFF, which was evaluated for IgAN in the BRIGHT-SC trial (n = 57), an international multi-center, randomized, double-blind, placebo-controlled phase 2/3 study (NCT02062684), the results of which are awaited.
Therapies directed against APRIL include the zigakibart (BION-1301) and sibeprenlimab (VIS649). Preliminary results from the phase 1/2 trial of zigakibart show that APRIL inhibition is associated with both reductions in proteinuria and circulating Gd-IgA1 levels.141 A phase 3 trial evaluating zigakibart in IgAN is currently underway (BEYOND; NCT05852938). The phase 2 trial of sibeprenlimab equally demonstrated that APRIL inhibition suppresses serum IgA and Gd-IgA1 and reduces proteinuria (ENVISION; NCT04287985).142 Sibeprenlimab is currently being evaluated in a phase 3 study (VISIONARY; NCT05248646).
Atacicept (VT-001), telitacicept, and povetacicept (ALPN-303) are TACI fusion proteins capable of inhibiting both BAFF and APRIL simultaneously. Atacicept (VT-001) has been evaluated in two phase 2 placebo-controlled RCTs (JANUS, NCT02808429, and ORIGIN, NCT04716231). In both studies, atacicept suppressed Gd-IgA1 levels and reduced proteinuria compared with placebo.143 Telitacicept has been evaluated in a phase 2 trial in China (NCT04905212), where it produced a 49% reduction in mean proteinuria and stabilized eGFR.144 A further phase 3 study is planned, aiming to recruit over 300 patients with IgAN (NCT05799287). Povetacicept (ALPN-303) is currently being assessed in the RUBY-3 trial, an open-label phase 1 and 2 study in 40 adults with autoimmune kidney diseases, including IgAN (NCT05732402).
Modulating the endothelin system
Endothelins are short peptides primarily produced by vascular endothelial cells, and glomerular endothelin-1 expression is associated with poorer outcomes in IgAN.145 Endothelin receptors are G-protein-coupled receptors constitutively expressed in human cells.146 These receptors exist as two isoforms, endothelin receptor type A (ETA-R) and endothelin receptor type B (ETB-R). ETA-R expression on vascular smooth muscle cells mediates vasoconstriction, while its expression on mesangial cells can mediate mesangial proliferation and inflammation:147 ETA-R antagonism in the ddY mouse model of IgAN suppresses severe histological changes and reduces proteinuria.148
Sparsentan is a dual ETA-R and angiotensin receptor antagonist, which has been evaluated in a phase 3 active-controlled RCT (PROTECT, NCT03762850). Interim analysis of PROTECT highlighted a nearly 49.8% reduction in proteinuria at 9 months, compared with a 15.1% reduction observed with the active control irbesartan, and 2-year eGFR data have recently been announced reporting improved preservation of kidney function with sparsentan compared with irbesartan.149 Sparsentan was also well tolerated. Sparsentan is the second drug approved by the FDA for use in IgAN at high risk of progression. Atrasentan, an ETA-R antagonist, is being evaluated in both a phase 2 study, which includes patients with IgAN (AFFINITY, NCT04573478), and a phase 3 study in IgAN (ALIGN, NCT04573478). Interim analysis of the AFFINITY study showed a 58.5% mean reduction in proteinuria among those with IgAN at 24 weeks.150
Conclusion
The contemporary understanding of the mechanisms driving IgAN has evolved considerably since it was first described. It is now clear that IgAN is a “multi-hit” disease and may involve perturbations in mucosal microbiomes, mucosal immunity, B cell and complement activation, as well as local inflammatory and fibrotic responses. Genetic and epigenetic controllers, influenced by environmental factors, appear to influence these mechanisms, perhaps explaining the ethnic differences in IgAN incidence and severity. These advances in the understanding of IgAN pathophysiology, combined with fundamental changes to the framework within which IgAN trials can be conducted, have prompted a surge in therapies being evaluated for this common cause of glomerular disease, and we have already seen the first two drugs receive approval for use in IgAN. This is an exciting time in the history of IgAN, with the hope that, in the coming years, we will have multiple treatment options for our patients. Building on the success of existing clinical trials is essential, with the inclusion of children with IgAN, patients with recurrent IgAN in their allograft, and children and adults with IgA vasculitis being a key priority.
Conflicts of interest
There are no conflicts of interest.
References
- An overview of regular dialysis treatment in Japan (as of 31 December 2004) Ther Apher Dial. 2006;10:476-97.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology, impact and preventive care of chronic kidney disease in Taiwan. Nephrology. 2010;15:3-9.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term outcomes for primary glomerulonephritis: New Zealand glomerulonephritis study. Nephrology. 2015;20:899-907.
- [CrossRef] [PubMed] [Google Scholar]
- Immunoglobulin a nephropathy: A pathophysiology view. Inflamm Res. 2016;65:757-70.
- [CrossRef] [PubMed] [Google Scholar]
- The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22:1795-803.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- IgA nephropathy. Clin J Am Soc Nephrol. 2017;12:677-86.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Long-term outcomes in IgA nephropathy. Clin J Am Soc Nephrol. 2023;18:727-38.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Epidemiology of IgA nephropathy: A global perspective. Semin Nephrol. 2018;38:435-42.
- [CrossRef] [PubMed] [Google Scholar]
- New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015;88:974-89.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- IgA nephropathy genetic risk score to estimate the prevalence of IgA nephropathy in UK biobank. Kidney Int Rep. 2020;5:1643-50.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43:321-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The role of complement in IgA nephropathy. Mol Immunol. 2019;114:123-32.
- [CrossRef] [PubMed] [Google Scholar]
- Immunological drivers of IgA nephropathy: Exploring the mucosa–kidney link. Int J Immunogenet. 2022;49:8-21.
- [CrossRef] [PubMed] [Google Scholar]
- Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol. 2010;28:243-73.
- [CrossRef] [PubMed] [Google Scholar]
- B Cell class switching in intestinal immunity in health and disease. Scand J Immunol. 2022;95:e13139.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Structure and function relationships in IgA. Mucosal Immunol. 2011;4:590-7.
- [CrossRef] [PubMed] [Google Scholar]
- IgA Function--variations on a theme. Immunology. 2004;113:175-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Bacterial IgA protease-mediated degradation of agigA1 and agIgA1 immune complexes as a potential therapy for IgA nephropathy. Sci Rep. 2016;6:30964.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The origin and activities of IgA1-containing immune complexes in IgA nephropathy. Front Immunol. 2016;7:117. doi: 10.3389/fimmu. 2016.00117
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007;71:1148-54.
- [CrossRef] [PubMed] [Google Scholar]
- New insights into the pathogenesis of IgA nephropathy. Pediatr Nephrol. 2018;33:763-77.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol. 2008;19:1008-14.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Genetic studies of IgA nephropathy: Past, present, and future. Pediatr Nephrol. 2010;25:2257-68.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Galactosylation of IgA1 is associated with common variation in C1GALT1. J Am Soc Nephrol. 2017;28:2158-66.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- O-glycosylation of serum IgD in IgA nephropathy. J Am Soc Nephrol. 2006;17:1192-9.
- [CrossRef] [PubMed] [Google Scholar]
- Aberrant Galactosylation of IgA1 is involved in the genetic susceptibility of chinese patients with IgA nephropathy. Nephrol Dial Transplant. 2009;24:3372-5.
- [CrossRef] [PubMed] [Google Scholar]
- Somatic mutations modulate autoantibodies against galactose-deficient IgA1 in IgA nephropathy. J Am Soc Nephrol. 2016;27:3278-84.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol. 2012;23:1579-87.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- IgA glycosylation and IgA immune complexes in the pathogenesis of IgA Nephropathy. Semin Nephrol. 2008;28:78-87.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Serum levels of galactose-deficient immunoglobulin (Ig) A1 and related immune complex are associated with disease activity of IgA nephropathy. Clin Exp Nephrol. 2014;18:770-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Poly-IgA complexes and disease severity in IgA nephropathy. Clin J Am Soc Nephrol. 2021;16:1652-64.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Protective role of IgA1 glycans against IgA1 self-aggregation and adhesion to extracellular matrix proteins. J Am Soc Nephrol. 1998;9:2048-54.
- [CrossRef] [PubMed] [Google Scholar]
- Role of IgA receptors in the pathogenesis of IgA nephropathy. J Nephrol. 2016;29:5-11.
- [CrossRef] [PubMed] [Google Scholar]
- Crosslinking of the human Fc receptor for IgA (FcalphaRI/CD89) Triggers FcR Gamma-chain-dependent shedding of soluble CD89. J Immunol. 1999;163:5806-12.
- [PubMed] [Google Scholar]
- Fcalpha receptor (CD89) mediates the development of immunoglobulin a (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J Exp Med. 2000;191:1999-2009.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med. 2012;209:793-806.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- β1,4-galactosyltransferase 1 is a novel receptor for IgA in human mesangial cells. Kidney Int. 2017;92:1458-68.
- [CrossRef] [PubMed] [Google Scholar]
- Deposition of IgA in primary IgA nephropathy: It takes at least four to Tango. Nephrol Dial Transplant. 2012;28:794-7.
- [CrossRef] [PubMed] [Google Scholar]
- Spleen tyrosine kinase inhibition ameliorates tubular inflammation in IgA nephropathy. Front Physiol. 2021;12:650888. doi: 10.3389/ fphys. 2021.650888
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Identification of IgA autoantibodies targeting mesangial cells redefines the pathogenesis of IgA nephropathy. Sci Adv. 2023;9:eadd6734.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Prevalence, characteristics, and outcomes of incidental IgA glomerular deposits in donor kidneys. Kidney Int Rep. 2020;5:1914-24.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Histologic classification of IgA nephropathy: Past, present, and future. Semin Nephrol. 2018;38:477-84.
- [CrossRef] [PubMed] [Google Scholar]
- IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67:504-13.
- [CrossRef] [PubMed] [Google Scholar]
- Podocyte injury induced by mesangial-derived cytokines in IgA nephropathy. Nephrol Dial Transplant. 2008;24:62-72.
- [CrossRef] [PubMed] [Google Scholar]
- Podocytopathy in the mesangial proliferative immunoglobulin a nephropathy: New insights into the mechanisms of damage and progression. Nephrol Dial Transplant. 2019;34:1280-5.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular insights into the pathogenesis of IgA nephropathy. Trends Mol Med. 2015;21:762-75.
- [CrossRef] [PubMed] [Google Scholar]
- Spleen tyrosine kinase is important in the production of proinflammatory cytokines and cell proliferation in human mesangial cells following stimulation with IgA1 isolated from IgA nephropathy patients. J Immunol. 2012;189:3751-8.
- [CrossRef] [PubMed] [Google Scholar]
- Correlation of disease activity in proliferative glomerulonephritis with glomerular spleen tyrosine kinase expression. Kidney Int. 2015;88:52-60.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- IgA1 immune complexes from pediatric patients with IgA nephropathy activate cultured human mesangial cells. Nephrol Dial Transplant. 2011;26:3451-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Synergistic effect of mesangial cell-induced CXCL1 and TGF-β1 in promoting podocyte loss in IgA nephropathy. PLoS One. 2013;8:e73425. doi: 10.1371/ journal.pone. 0073425
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Serum IgA1 from Patients with IgA nephropathy up-regulates integrin-linked kinase synthesis and inhibits adhesive capacity in podocytes through indirect pathways. Clin Invest Med. 2009;32:E20-7.
- [CrossRef] [PubMed] [Google Scholar]
- Evidence from the oxford classification cohort supports the clinical value of subclassification of focal segmental glomerulosclerosis in IgA nephropathy. Kidney Int. 2017;91:235-43.
- [CrossRef] [PubMed] [Google Scholar]
- Proteinuria: Detection and role in native renal disease progression. Transplant Rev (Orlando). 2012;26:3-13.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular insight in intrarenal inflammation affecting four main types of cells in nephrons in IgA nephropathy. Front Med (Lausanne). 2023;10:1128393. doi: 10.3389/fmed. 2023.1128393
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Activation of tubular epithelial cells by mesangial-derived TNF-alpha: glomerulotubular communication in IgA nephropathy. Kidney Int. 2005;67:602-12.
- [CrossRef] [PubMed] [Google Scholar]
- The role of mononuclear phagocyte system in IgA nephropathy: Pathogenesis and prognosis. Front Immunol. 2023;14
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Macrophage interactions with collecting duct epithelial cells are capable of driving tubulointerstitial inflammation and fibrosis in immunoglobulin a nephropathy. Nephrol Dial Transplant. 2020;35:1865-77.
- [CrossRef] [PubMed] [Google Scholar]
- Complement in autoimmune diseases. Clin Chim Acta. 2017;465:123-30.
- [CrossRef] [PubMed] [Google Scholar]
- Essential role of complement in pregnancy: from implantation to parturition and beyond. Front Immunol. 2020;11:1681. doi: 10.3389/fimmu. 2020.01681
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Predictive value of mesangial C3 and C4d deposition in IgA nephropathy. Clin Immunol. 2020;211:108331. doi: 10.1016/j.clim. 2019.108331
- [CrossRef] [PubMed] [Google Scholar]
- complement membrane attack (MAC) in idiopathic IgA-glomerulonephritis. Kidney Int. 1987;31:820-9.
- [CrossRef] [PubMed] [Google Scholar]
- The immunohistology of IgA nephropathy. Am J Kidney Dis. 1988;12:348-52.
- [CrossRef] [PubMed] [Google Scholar]
- Composition of mesangial deposits in IgA nephropathy: Complement factors. Nephron. 1987;46:219.
- [CrossRef] [PubMed] [Google Scholar]
- Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol. 2015;26:1503-12.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Complement activation in IgA nephropathy. Semin Immunopathol. 2021;43:679-90.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Activation of the alternative pathway of complement by human serum IgA. Eur J Immunol. 1987;17:321-6.
- [CrossRef] [PubMed] [Google Scholar]
- Complement-fixing properties of human IgA antibodies. Alternative pathway complement activation by plastic-bound, but not specific antigen-bound, IgA. Scand J Immunol. 1989;30:175-83.
- [CrossRef] [PubMed] [Google Scholar]
- Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol. 2006;17:1724-34.
- [CrossRef] [PubMed] [Google Scholar]
- Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol. 2001;167:2861-8.
- [CrossRef] [PubMed] [Google Scholar]
- Immunopathogenesis of IgAN. Semin Immunopathol. 2007;29:427-43.
- [CrossRef] [PubMed] [Google Scholar]
- The role of IgA in the pathogenesis of IgA nephropathy. Int J Mol Sci. 2019;20:6199. doi: 10.3390/ ijms20246199
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The role of secretory IgA and complement in IgA nephropathy. Semin Nephrol. 2008;28:58-65.
- [CrossRef] [PubMed] [Google Scholar]
- The level of serum secretory IgA of patients with IgA nephropathy is elevated and associated with pathological phenotypes. Nephrol Dial Transplant. 2008;23:207-12.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261-92.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Enhancement of polymeric immunoglobulin receptor transcytosis by biparatopic VHH. PLoS One. 2011;6:e26299.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Steady-state generation of mucosal IgA+ plasmablasts is not abrogated by B-cell depletion therapy with rituximab. Blood. 2010;116:5181-90.
- [CrossRef] [PubMed] [Google Scholar]
- A randomized, controlled trial of rituximab in IgA nephropathy with proteinuria and renal dysfunction. J Am Soc Nephrol. 2017;28:1306-13.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Associations of genetic variants contributing to gut microbiota composition in immunoglobin a nephropathy. mSystems. 2021;6:e00819-20. doi: 10.1128/mSystems. 00819-20
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Modifications of gut microbiota are associated with the severity of IgA nephropathy in the chinese population. Int Immunopharmacol. 2020;89:107085. doi: 10.1016/j.intimp. 2020.107085
- [CrossRef] [PubMed] [Google Scholar]
- The potential role of an aberrant mucosal immune response to SARS-CoV-2 in the pathogenesis of IgA nephropathy. Pathogens. 2021;10:881. doi: 10.3390/pathogens10070881
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Salivary microbiota associated with immunoglobulin a nephropathy. Microb Ecol. 2015;70:557-65.
- [CrossRef] [PubMed] [Google Scholar]
- Gut microbiota in immunoglobulin a nephropathy: A malaysian perspective. BMC Nephrol. 2021;22:145.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Microbiota and metabolome associated with immunoglobulin a nephropathy (IgAN) PLoS One. 2014;9:e99006. doi: 10.1371/journal.pone. 0099006
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- A comparative study of the gut microbiota associated with immunoglobulin a nephropathy and membranous nephropathy. Front Cell Infect Microbiol. 2020;10:557368. doi: 10.3389/fcimb. 2020.557368
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Exaggerated systemic antibody response to mucosal helicobacter pylori infection in IgA nephropathy. Am J Kidney Dis. 1999;33:1049-57.
- [CrossRef] [PubMed] [Google Scholar]
- O-glycosylation of serum IgA1 antibodies against mucosal and systemic antigens in IgA nephropathy. J Am Soc Nephrol. 2006;17:3520-8.
- [CrossRef] [PubMed] [Google Scholar]
- Mice overexpressing BAFF develop a commensal flora–dependent, IgA-associated nephropathy. J Clin Investig. 2011;121:3991-4002.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- TLR9 and BAFF: Their expression in patients with IgA nephropathy. Mol Med Rep. 2014;10:1469-74.
- [CrossRef] [PubMed] [Google Scholar]
- Dendritic cells, BAFF, and APRIL: Innate players in adaptive antibody responses. Immunity. 2002;17:235-8.
- [CrossRef] [PubMed] [Google Scholar]
- Systematic review of safety and efficacy of atacicept in treating immune-mediated disorders. Front Immunol. 2020;11:433. doi: 10.3389/fimmu. 2020.00433
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Toll-like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol. 2008;19:2384-95.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Association between toll-like receptor 10 (TLR10) gene polymorphisms and childhood IgA nephropathy. Eur J Pediatr. 2011;170:503-9.
- [CrossRef] [PubMed] [Google Scholar]
- Toll-like receptors, immunoproteasome and regulatory T cells in children with henoch-schönlein purpura and primary IgA nephropathy. Pediatr Nephrol. 2014;29:1545-51.
- [CrossRef] [PubMed] [Google Scholar]
- Increased APRIL expression induces IgA1 aberrant glycosylation in IgA nephropathy. Medicine. 2016;95:e3099. doi: 10.1097/ MD.0000000000003099
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- External suppression causes the low expression of the cosmc gene in IgA nephropathy. Nephrol Dial Transplant. 2008;23:1608-14.
- [CrossRef] [PubMed] [Google Scholar]
- Serum BAFF is elevated in patients with IgA nephropathy and associated with clinical and histopathological features. J Nephrol. 2013;26:683-90.
- [CrossRef] [PubMed] [Google Scholar]
- Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46:1187-96.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Global incidence of IgA nephropathy by race and ethnicity: A systematic review. Kidney360. 2023;4:1112-2.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Individuals of pacific asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney Int. 2013;84:1017-24.
- [CrossRef] [PubMed] [Google Scholar]
- GWAS defines pathogenic signaling pathways and prioritizes drug targets for IgA nephropathy. medRxiv 2021 2021.11.19.21265383. doi: 10.1101/2021.11.19.21265383
- [CrossRef] [PubMed] [Google Scholar]
- Genome-wide association analyses define pathogenic signaling pathways and prioritize drug targets for IgA nephropathy. Nat Genet. 2023;55:1091-105.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97.
- [CrossRef] [PubMed] [Google Scholar]
- MicroRNAs in kidney physiology and disease. Nat Rev Nephrol. 2015;11:23-33.
- [CrossRef] [PubMed] [Google Scholar]
- MicroRNAs in chronic kidney disease: Four candidates for clinical application. Int J Mol Sci. 2020;21:6547. doi: 10.3390/ ijms21186547
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- An overview of MicroRNAs: Biology, functions, therapeutics, and analysis methods. J Cell Physiol. 2019;234:5451-65.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical application of MicroRNAs in glomerular diseases. Nephrol Dial Transplant. 2022;38:1375-84.
- [CrossRef] [PubMed] [Google Scholar]
- In a retrospective international study, circulating miR-148b and let-7b were found to be serum markers for detecting primary IgA nephropathy. Kidney Int. 2016;89:683-92.
- [CrossRef] [PubMed] [Google Scholar]
- Serum levels of miR-148b and Let-7b at diagnosis may have important impact in the response to treatment and long-term outcome in IgA nephropathy. J Clin Med. 2021;10:1987. doi: 10.3390/jcm10091987
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Abnormal miR-148b expression promotes aberrant glycosylation of IgA1 in IgA nephropathy. J Am Soc Nephrol. 2012;23:814-24.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Role of let-7b in the regulation of N-acetylgalactosaminyltransferase 2 in IgA nephropathy. Nephrol Dial Transplant. 2015;30:1132-9.
- [CrossRef] [PubMed] [Google Scholar]
- Urinary sediment miRNAs reflect tubulointerstitial damage and therapeutic response in IgA nephropathy. BMC Nephrol. 2017;18:63.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Kidney MicroRNA-21 expression and kidney function in IgA Nephropathy. Kidney Med. 2021;3:76-82.e1.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- MicroRNA-21: A central regulator of fibrotic diseases via various targets. Curr Pharm Des. 2015;21:2236-42.
- [CrossRef] [PubMed] [Google Scholar]
- Evaluating a new international risk-prediction tool in IgA nephropathy. JAMA Intern Med. 2019;179:942-52.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- A pilot study to predict risk of IgA nephropathy progression based on miR-204 expression. Kidney Int Rep. 2021;6:2179-88.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Differential expression of MicroRNA miR-150-5p in IgA nephropathy as a potential mediator and marker of disease progression. Kidney International. 2021;99:1127-39.
- [CrossRef] [PubMed] [Google Scholar]
- The MicroRNA miR-155 is essential in fibrosis. Non-Coding RNA. 2019;5:23. doi: 10.3390/ncrna5010023
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Potential role of upregulated MicroRNA-146b and -21 in renal fibrosis. Mol Med Rep. 2017;16:2863-7.
- [CrossRef] [PubMed] [Google Scholar]
- MiR-135a promotes renal fibrosis in diabetic nephropathy by regulating TRPC1. Diabetologia. 2014;57:1726-36.
- [CrossRef] [PubMed] [Google Scholar]
- Renal microRNA- and RNA-profiles in progressive chronic kidney disease. Eur J Clin Investig. 2016;46:213-26.
- [CrossRef] [PubMed] [Google Scholar]
- MicroRNA-204-5p Suppresses IL6-mediated inflammatory response and chemokine generation in HK-2 renal tubular epithelial cells by targeting IL6R. Biochem Cell Biol. 2018;97:109-17.
- [PubMed] [Google Scholar]
- Proteinuria reduction as a surrogate end point in trials of IgA nephropathy. Clin J Am Soc Nephrol. 2019;14:469-81.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- IgA nephropathy: An overview of drug treatments in clinical trials. Expert Opin Investig Drugs. 2022;31:1321-38.
- [CrossRef] [PubMed] [Google Scholar]
- Current knowledge of targeted-release budesonide in immunoglobulin A nephropathy: A comprehensive review. Front Immunol. 2023;13:926517. doi: 10.3389/fimmu. 2022.926517
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- FC050: Nefecon® selectively modifies the composition of circulating immune complexes in IGA nephropathy. Nephrol Dial Transplant. 2022;37(Suppl 3):i815-7.
- [CrossRef] [Google Scholar]
- P0344NEFECON® (budesonide) selectively reduces circulating levels of BAFF (BLYS) and soluble BCMA and TACI in IGA nephropathy. Nephrol Dial Transplant. 2020;35(Suppl 3) doi: 10.1093/ndt/gfaa142.P0344
- [CrossRef] [Google Scholar]
- Results from part A of the multi-center, double-blind, randomized, placebo-controlled NefIgArd trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin a nephropathy. Kidney Int. 2023;103:391-402.
- [CrossRef] [PubMed] [Google Scholar]
- POS-830 Nefecon for the treatment of IgA nephropathy in patients at Risk of progressing to end-stage renal disease: The nefigard phase 3 trial results. Kidney Int Rep. 2021;6:S361.
- [CrossRef] [Google Scholar]
- Efficacy and safety of a targeted-release formulation of budesonide in patients with primary IgA nephropathy (NefIgArd): 2-year results from a randomised phase 3 Trial. Lancet. 2023;402:859-70.
- [CrossRef] [PubMed] [Google Scholar]
- Fecal microbiota transplantation: In perspective. Therap Adv Gastroenterol. 2016;9:229-39.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Alleviation of refractory IgA nephropathy by intensive fecal microbiota transplantation: The first case reports. Ren Fail. 2021;43:928-33.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- POS-546 efficacy and safety of iptacopan in IgA nephropathy: Results of a randomized double-blind placebo-controlled phase 2 study at 6 months. Kidney International Reports. 2022;7:S236.
- [CrossRef] [Google Scholar]
- Alternative complement pathway inhibition with iptacopan for the treatment of C3 glomerulopathy-study design of the APPEAR-C3G trial. Kidney Int Rep. 2022;7:2150-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- POS-107 long-term phase 2 efficacy of the MASP-2 inhibitor narsoplimab for treatment of severe IGA nephropathy. Kidney Int Rep. 2022;7:S45.
- [CrossRef] [Google Scholar]
- C5a receptor inhibitor avacopan in immunoglobulin a nephropathy-an open-label pilot study. Clin Kidney J. 2022;15:922-8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- New treatment strategies for IgA nephropathy: Targeting plasma cells as the main source of pathogenic antibodies. J Clin Med. 2022;11
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- POS-109 interim results of phase 1 and 2 trials to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics, and clinical activity of BION-1301 in patients with IgA nephropathy. Kidney Int Rep. 2022;7:S46.
- [CrossRef] [Google Scholar]
- WCN23-0684 interim biomarker analysis from a randomized, double-blind, placebo-controlled, phase 2 trial of sibeprenlimab (VIS649) in participants with immunoglobulin a nephropathy. Kidney Int Rep. 2023;8(3, Supplement):S76-7.
- [CrossRef] [Google Scholar]
- Randomized phase ii JANUS study of atacicept in patients with IgA nephropathy and persistent proteinuria. Kidney Int Rep. 2022;7:1831-41.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Randomized phase 2 trial of telitacicept in patients with IgA nephropathy with persistent proteinuria. Kidney Int Rep. 2023;8:499-506.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Renal endothelin-1 and endothelin receptor type B expression in glomerular diseases with proteinuria. J Am Soc Nephrol. 2001;12:2321-9.
- [CrossRef] [PubMed] [Google Scholar]
- Endothelin system: The double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851-76.
- [CrossRef] [PubMed] [Google Scholar]
- POS-370 human renal mesangial cell activation induced by endothelin-1 or IGA nephropathy patient-derived immune complexes is blocked by selective eta antagonist atrasentan. Kidney Int Rep. 2021;6 (4, Suppl):S160-1.
- [CrossRef] [Google Scholar]
- Effect of a specific endothelin receptor a antagonist on glomerulonephritis of ddY mice with IgA nephropathy. Nephron. 1996;72:454-60.
- [CrossRef] [PubMed] [Google Scholar]
- Sparsentan in patients with IgA nephropathy: A prespecified interim analysis from a randomised, double-blind, active-controlled clinical trial. Lancet. 2023;401:1584-94.
- [PubMed] [Google Scholar]
- FC052: atrasentan for the treatment of IGA nephropathy: interim results from the affinity study. nephrol dial transplant. 2022;37 doi: 10.1093/ndt/gfac107.004
- [CrossRef] [Google Scholar]




